I am looking for an existing SWM4-NDP polarizable water model file in (.ff/.zmat/.xyz format) suitable for LAMMPS Drude simulations, preferably compatible with CL&Pol-style workflows.
I would greatly appreciate any help or references.
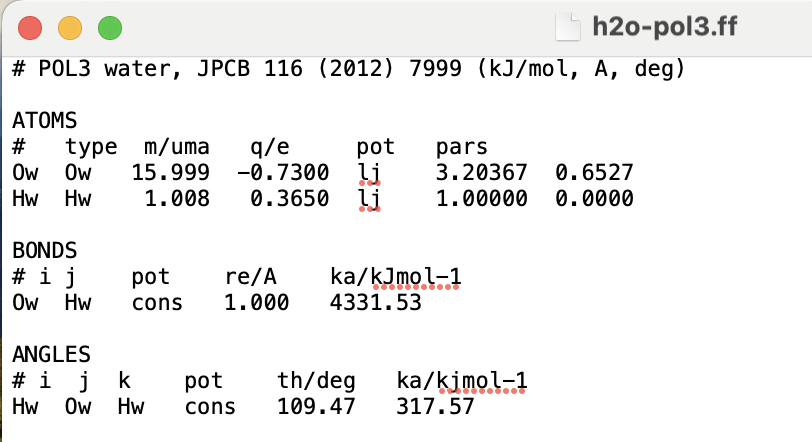
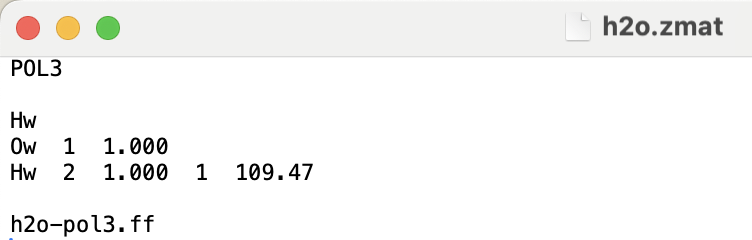
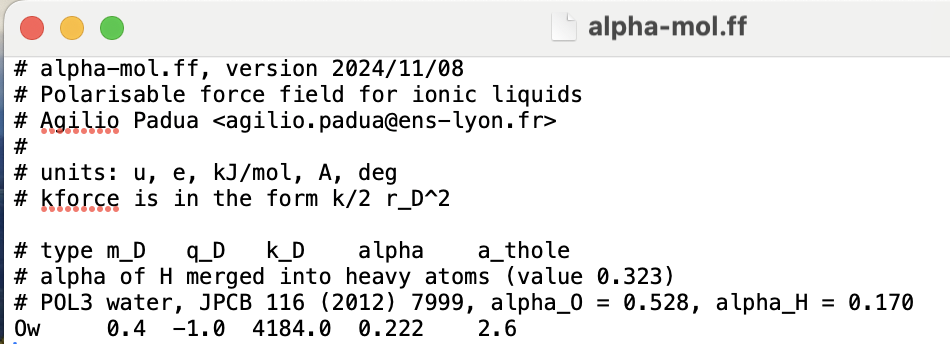
To clarify what I meant by “CL&Pol-style workflow”: I am attaching the fragment-style definition we use for POL3 water in our simulations. It consists of:
a modular .ff file defining atom types and LJ parameters,
a .zmat file defining molecular geometry,
an alpha-mol.ff entry defining the Drude parameters.
This allows us to build mixed systems (electrolytes + water) using fragment assembly tools and then apply controlled LJ cross-interaction scaling between fragments.
The SWM4-NDP example in LAMMPS package is a complete demonstration system, but not a reusable fragment-style definition of the model.
My question is whether an SWM4-NDP parameterization exists in a similarly modular format (force-field fragment + Drude definition), or whether extracting and rebuilding it from the example input is the intended workflow.
The general approach from the LAMMPS developers is that system preparation is an external task to be done with specialized tools and then - if needed - the files are converted to what LAMMPS needs. This is mostly due to the fact that LAMMPS is so extremely flexible, that it is impossible for the LAMMPS developers to have a toolkit available and validate it for the many different applications. We focus on the “simulation engine” parts. That said, we bundle some contributed tools in the tools folder. But those tend to become obsolete faster than the main simulation sources and are often not very well maintained (or just set up for the specific needs of the person that contributed them).
In addition, you have to keep in mind that quite a chunk of LAMMPS features are contributed code, so that we often don’t know much about the workflows and the provide example files are all we have to confirm functionality. How well packages are maintained depends on many different factors. It goes beyond the scope of this discussion (and this forum) to elaborate on those.
In conclusion, what you are asking about is not something that the LAMMPS developers worry much about. This is rather of interest to people that work in that specific domain. Candidates for that are either the research group or groups that contributed that specific feature or groups that make significant use of it. Those likely have in-house tools and may have templates etc. All that we have is what is in the source code tarball or git repository.
While you might think that a forum like this one is where LAMMPS users hang out and share their successes and thoughts and tools - and it has always been our hope that this happens - the reality is far from it. For the most part we have questions with people struggling to make their inputs work or generally to understand how to run simulations or why they are crashing or not producing the results they expect.
I am in the “simulation business” now for more than 30 years and I have yet to see a research community where people are more paranoid and less welling to share and help others. In fact, I recall a long time ago being told by a supervisor that I should not help people outside the research group so much as I would only strengthen the competition and there is too much competition already.
To help achieve more modularity, here are some ideas and potential pitfalls:
You can define molecule templates and use LAMMPS’s create_atoms command to create molecules. These molecule templates can include Drude particles – there is nothing special about a Drude particle until a fix drude command “marks” them for special handling by integrators. This example for SPC/E water can be easily adapted to SWM4-NDP water: 10.4.8. SPC and SPC/E water model — LAMMPS documentation
It’s important to note that LAMMPS still expect to know exactly how many types there will be at the start of a simulation, when the box is being first defined. So you will still have to count types and declare boxes accordingly (see the dataset in the second dot point above for useful ways to navigate this complexity).
Finally, it is entirely feasible to instead use the customary Moltemplate ( https://moltemplate.org/ ) to prepare a LAMMPS data file, reading in coordinates constructed by Packmol. You can write your own Moltemplate force field file reasonably easily.