Hi Axel and Steve,
Thanks for taking the time with me on this. I did speak to my supervisor on
this be coming to the group but he has no experience with parallel systems,
this is our first attempt. Basically he said if I can’t sort it all out
today to just go serial so everything will have a local ID and come back to
parallel in a couple of months.
sorry, but that is bad advice. LAMMPS is designed from the ground up
to be parallel and even in serial mode it uses a parallel library
"stub" to be able to use the same code base. furthermore, the advice
assumes that LAMMPS way to handle coordinate follows minimum image
conventions, which is not correct. so even in serial mode, you can
have multiple copies of the same atom around. using Atom::map() will
give you only one of them, usually the one that is owned by the
domain. yet, that may not be the one you need, if one or more of your
tuple of angle atoms crosses a domain boundary.
if you ignore the basic management strategies in LAMMPS, you can
indeed get a serial code working, but you will have to spend time to
program features that you need which would otherwise automatically be
provided by LAMMPS and on top of that, you will have a *much* harder
time to make it work in parallel, because you will mostly have to undo
and then properly redo what you did for the serial version, as that
will not work in parallel (or will kill performance).
The MB model is different as we only have a single bond arm per molecule,
not all exponentials are Gaussians, the LJ term is gone and being hybrided
with another custom LJ. i-1 and i+1 are not potential bonding arms, but
merely residues bonded to i. i and j are arbitrary. The is no torque term
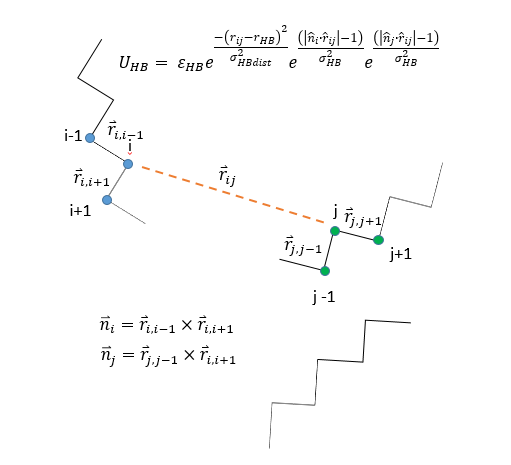
at all. I’ve attached a schematic of the situation.
ok. so i would say that calling this an MB model is a bit of a
misnomer, but that is besides the point of the discussion.
if i read the picture correctly, you have an additional energy term
between pairs of topological angles that depends on the distance
between the pairs of atoms i and j and the relative orientation of the
vector connecting the pair to the plane formed by the three atoms
comprising the respective angles (BTW: there seems to be a typo in the
definition of n_j).
this is indeed best implemented as a pair style to be used with
pair_style hybrid/overlay to add it to the regular interaction. as far
as i can see, there are a number of challenges to get this to work
well in LAMMPS (and that is mostly regardless of whether you try to
make it work in parallel or not).
1) are only r_ij pairs considered between different molecules or can
they also be in the same molecule? consider the case of a very long
chain: if this bends around, it will behave pretty much as if it was a
different molecule. in this case, one would have to determine what
would be a minimum distance between atom i and j on the same chain.
with the default exclusion settings (lj/coul 0.0 0.0 0.0) i,j pairs in
the same molecule will only show up in the neighborlist (or have a
non-zero scaling factor), if j is at least 4 atoms away from i, i.e.
there is one atom in between the two angles. this is probably not a
big deal in the beginning, but you should keep it in mind.
2) you have to take care of the different cases for the "newton" flag,
which can be on/off for both non-bonded and bonded interactions. this
affects what information is stored on ghost atoms and whether
contributions to force and energy need to be stored on ghost atoms.
the default of LAMMPS is "on" for both flags, but in your case, the
situation is likely going to be much easier when you require newton
off for both non-bonded pair and bonded interactions. programming for
the other cases can be done later.
3) it is indeed quite likely that would improve performance by
precomputing the normals for each atom triple before doing the force
loop. you can store it with the corresponding atom and then use a
forward communication to communicate it to its respective ghost atom
copies (either on the same or other processors). as an example, you
could look at the pair_eam.cpp file, where a single property (fp) is
stored with each atom. it is easy to extend this to have multiple
properties in there, you just need to adapt the
pack/unpack_forward_comm() functions accordingly. it may be simpler,
though, to first implement this without this optimization. once you
have a pair style that works correctly, you can consider all kinds of
optimizations but you will always have a reference to compare to.
(p.s.: you'll be having some serious "fun" deriving forces for this
potential...).
4) there are several optional methods that LAMMPS requires for special
cases (distance based r-RESPA, group/group energy/force computation
and more). these are implemented in many pair styles, but are not
required for running basic MD. so it is better to postpone those (e.g.
compute_inner/middle/outer, single) for later.
5) identifying the neighboring atoms should not make any assumptions
of the tag numbers being ordered or local atom indexing being done in
order. steve already mentioned that atom->map() will give you one of
the possible local/ghost indices, but that may not be the one you
need. that mapping also may change due to atoms migrating between
domains and local atoms being stored in a spatially sorted fashion
(for better performance). this happens during the re-neighboring
steps. more details are in steve's e-mail, so i won't repeat them
here.
in general, this is not a trivial model to add to a large package
program and i am a bit shocked by the rather cavalier attitude of your
adviser. there is so much badly written code floating around that is
causing increasing amounts of headaches to people, spending the time
to sufficiently understand the flow of control in a package program
requires time and some experimentation, but it is fully worth the
time, since afterwards you'll benefit a lot from the available
infrastructure and have something that is much easier to maintain and
improve.
HTH,
axel.