Dear lammps user,
my lammps version is 7 August 2019.
First, I hope my message is clear enough.
I am trying to do NVE simulation for two-dimensional Lennard Jones fluids composed of 121 atoms. I want the NVE ensemble to have a constant total energy corresponding to the total energy that the same system can have at temperature 0.48 .
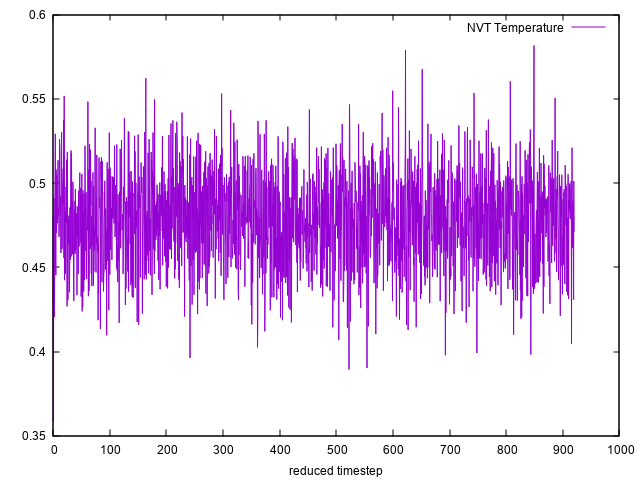
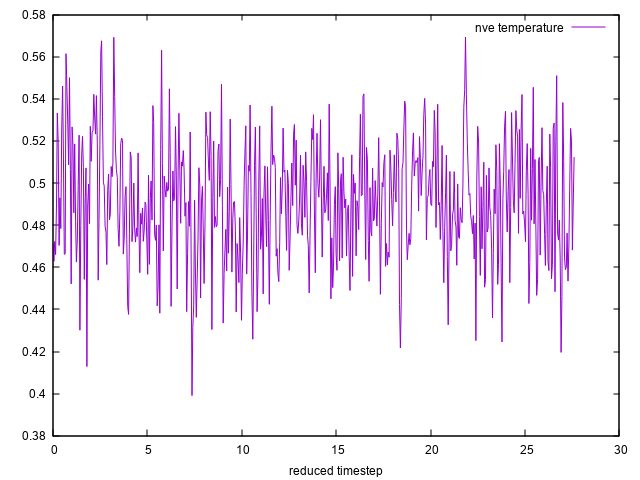
Therefore, I equilibrated my system by running first NVT simulation at temperature 0.48 up to 200000 timesteps. Then I immediately started doing NVE simulation up to 6000 timesteps for the same system after it had already been equilibrated by the NVT simulations.
I know very well that in the NVE ensemble the temperature is not constant. However, if I am not wrong, the temperature in the NVE ensemble should fluctuate around some value at equilibrium. Hence, I want this value of temperature to be 0.48.
I plotted the temperature during the NVT equilibration and during the NVE simulation and I attached the tow plots to this email. From the attached two plots, I can notice that the temperature in the NVE simulation does not fluctuate around 0.48 which means that the system has total energy different from that total energy obtained at temperature 0.48 . This will affect the plot of the radial distribution function.
Could you please help me understand the mistake I made ?. In the end, I want to plot the radial distribution function for the NVE simulation whose temperature fluctuate around 0.48 at equilibrium.
Below is the script I am using. I would like to say that there are fixed quantities that I have to follow according to some reference and they are : the timestep value, the temperature, the number of particles, the cutoff distance (6.5) and the number of timesteps in the NVE run command (6000).