Dear all,
I am currently working on a project to simulate a Silicon Nitride (SiN) system using LAMMPS with Tersoff potentials. I have set up my LAMMPS input script and created a Tersoff potential file for Si and N interactions, based on values from a paper (DOI: Hydrogen role on the properties of amorphous silicon nitride | Journal of Applied Physics | AIP Publishing).
Here is my LAMMPS input script:
# Initialize simulation
units metal
dimension 3
boundary p p p
atom_style full
# Create atoms
lattice diamond 5.43
region box block 0 2 0 2 0 2
create_box 2 box # Define 2 atom types (Si and N)
create_atoms 1 box # Create Si atoms in the box
create_atoms 2 box # Create N atoms in the box
# Define mass for atom types
mass 1 28.09 # Silicon
mass 2 14.01 # Nitrogen
# Define potential
pair_style tersoff
pair_coeff * * SiN.tersoff Si N
# Perform a calculation
run 100
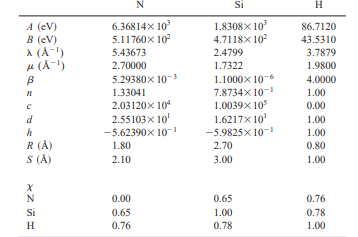
My Tersoff potential file is as follows:
# Tersoff potential for Si and N
# e1 e2 e3 m gamma lambda3 c d costheta0 n beta lambda2 B R D lambda1 A
Si Si Si 1.7322 1 2.4799 100390 1.6217 -0.59825 0.78734 0.0000011 0 471.18 2.7 0.3 2.4799 1830.8
Si Si N 1.7322 1 2.4799 100390 1.6217 -0.59825 0.78734 0.0000011 0 471.18 2.7 0.3 2.4799 1830.8
Si N Si 1.7322 1 2.4799 100390 1.6217 -0.59825 0.78734 0.0000011 0 471.18 2.7 0.3 2.4799 1830.8
Si N N 2.7 1 5.43673 20312 2.55103 -0.56239 1.33041 0.0052938 0 511.76 1.8 0.3 5.43673 6368.14
N Si Si 1.7322 1 2.4799 100390 1.6217 -0.59825 0.78734 0.0000011 0 471.18 2.7 0.3 2.4799 1830.8
N Si N 1.7322 1 2.4799 100390 1.6217 -0.59825 0.78734 0.0000011 0 471.18 2.7 0.3 2.4799 1830.8
N N Si 2.7 1 5.43673 20312 2.55103 -0.56239 1.33041 0.0052938 0 511.76 1.8 0.3 5.43673 6368.14
N N N 2.7 1 5.43673 20312 2.55103 -0.56239 1.33041 0.0052938 0 511.76 1.8 0.3 5.43673 6368.14
When I try to run the simulation, LAMMPS gives me an error: “Illegal Tersoff parameter (src/MANYBODY/pair_tersoff.cpp:510)”. I am running LAMMPS-omp 2022-06-23 on a Linux-based high-performance computing (HPC) cluster (the latest version we have).
Looking at my Tersoff potential file, the values for m, gamma, lambda2, and costheta0 parameters are identical for Si-Si, Si-N, and N-N interactions, which I am uncertain about. I could not find these specific parameters in the referenced paper.
Can anyone guide me on how to correctly format Tersoff potential parameters from the literature for use in LAMMPS please?
Thank you!
Brahim