Dear ATAT Community,
I am working on generating a Special Quasirandom Structure (SQS) for a LiPb alloy with (44.5% Pb, 55.5% Li) in an HCP crystal structure using the tool from ATAT. Despite following the documentation and an example from (Redirecting), I am encountering two issues and would appreciate your guidance.mcsqs
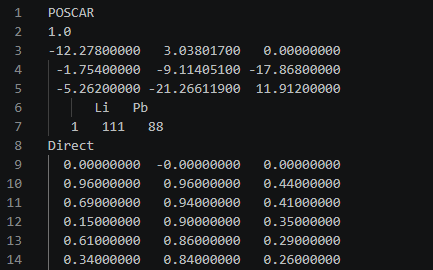
However, even when I set up supercells with 200 or 400 atoms, there is always one atom left without assigned atomic coordinates in the output .out file generated by calculation. I would like to ask what exactly causes this problem.
After converting the .out file to a POSCAR file, as shown in the figure, there is always one atom without assigned coordinates. Only 199 or 399 atomic coordinates are present in the file.
Hi,
maybe it will help to try to prepare the input file for mcsqs at https://simplysqs.com/ (or use the subsequent bash script that will also automatically convert the .out file format into POSCAR format, see also e.g. this tutorial https://www.youtube.com/watch?v=GGo_9T5wqus). As the input, just upload the initial hcp structure with your lattice parameters (e.g. alpha-Ti and change its lattice parameters to yours 3.508 3.508 5.956).
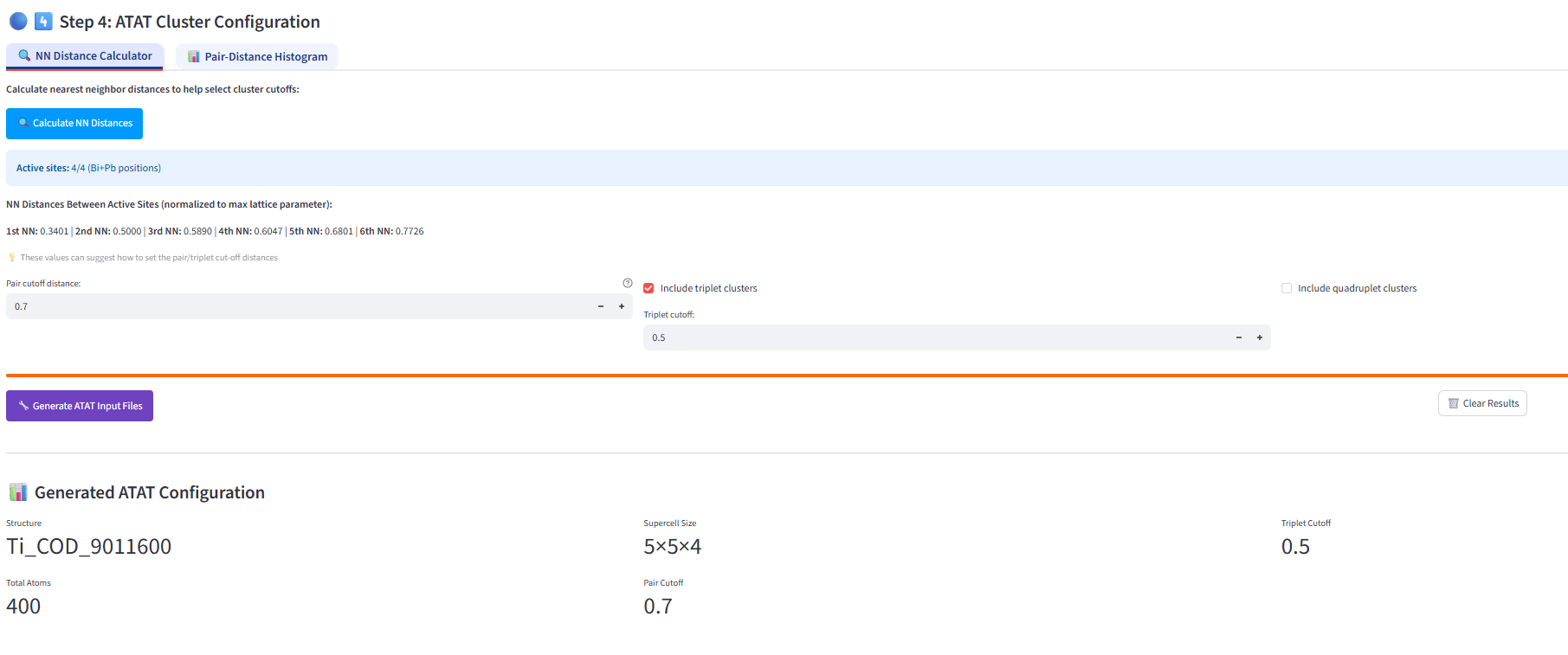
I have successfully completed ATAT-related calculations using SimplySQS. However, I have another question: how should I set the Pair cutoff distance and Triplet cutoff, and what is the minimum value of the Objective function that indicates a good model construction?It seems that no explicit specifications are given in the ATAT user manual.
I would suggest setting the cutoffs based on nearest-neighbor distances, for example using the distance up to the 4th nearest neighbor for pairs and up to the 2nd nearest neighbor for triplets.
I don’t think there is a universal minimum objective-function value that indicates a good SQS model. Generally, a lower value means a better match to the target random structure. I would recommend running several independent searches, for example 5 parallel runs, and selecting the SQS with the lowest objective function, while checking how the objective functions are changing with time.
I set the parameters as suggested, yet the calculated objective function value is only -0.9. In contrast, I previously obtained a value of -3.69 by running the command
the units of lattice parameters in the web interface are normalized to 1 (the largest lattice parameter is 1). This is better for me when comparing NN distances. The lattice parameters corresponding to your initial structure are added in postprocessing of the bestsqs.out (automatically done with the bash script by creating POSCAR or you can upload the bestsqs.out back into the interface).
With your previous settings, I see you considered the absolute lattice parameters, where your settings probably included much lower number of pairs and triplets combinations.