Dear all,
I am trying to perform a coarse-grain MD simulation of double strand DNA. The coarse-grain model is 3SPN2. I used implict solvent and ions and periodic boundary conditions under NVT ensemble. After the simulation was finished, I loaded the trajectory into VMD. Strangely, the DNA looked ok in the first hundred frames but thereafter some beads moved far from DNA chains, looking like being detached. However sometimes it was intact, just as follows:
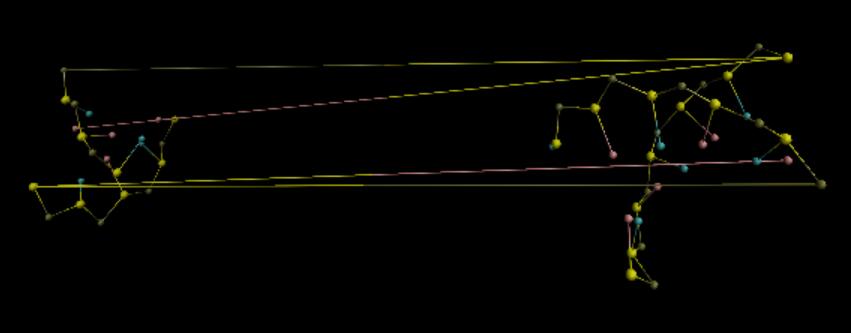
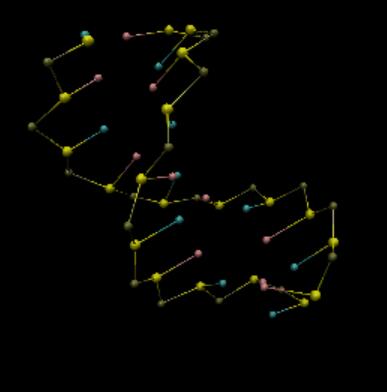
The box is:
-38.870000 38.870000 xlo xhi
-38.870000 38.870000 ylo yhi
-38.870000 38.870000 zlo zhi
I found that some coordinates of trajectory were larger than 38.87. In the docs of dump command, it notes that the coordinates of an atom written to a dump file may be slightly outside the simulation box. But I feel it’s abnormal in my case, because I think the beads should not have moved away from the DNA chain.
I’m very confused about this. Could you give me some guidance? I attached input and configuration file. Thank you very much in advance.
lmp.in (2.6 KB)
conf_lmp.in (7.2 KB)