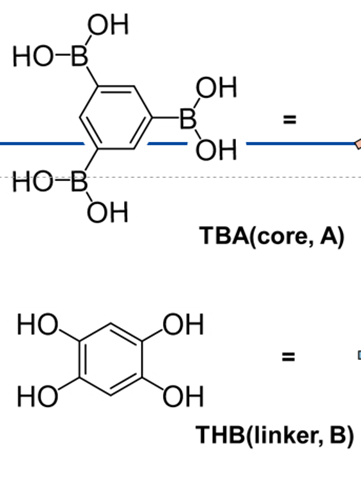
Hello everyone, when using LAMMPS’ bond/react to simulate a reversible reaction between molecules A and B, and the program encounters a ‘bond atom missing’ error. The reaction scheme as follows: Step 1 is the condensation of A and B to form boric acid plus two hydroxyl groups and release two water molecules. Steps 2 and 3 are two successive hydrolysis reactions that regenerate the original A and B molecules.
I have finished writing the templates for the three-step reaction, and when combined they successfully execute the forward and reverse reactions. However, although the reversible reaction now proceeds as expected, the simulation becomes unstable at later stages—around step 326,800 a “bond atom missing” error occurs, as shown below:
I have examined the trajectory and found that the missing bonds can occur at the water O–H, at C–H bonds in the solvent, or at C–H bonds within the A and B molecules, among others. The figure below shows an O–H bond in water that has gone missing.
I tried stabilizing the molecular energy by using fix 4 bond_react_MASTER_group temp/rescale 1 {T} {T} ${T} 1 1, but the “bond atom missing” error still keeps occurring. And input data as follow:
I’m very sorry, the data file is too large to upload. I’ve provided my email address—if you send me a message, I will immediately forward the data file to you. Thank you very much for your help. My email is [email protected].
Do you have an example with a simpler/smaller data file? That will be best for those trying to help you. If not, you can provide a link to download the file as well.
Thank you very much for your prompt response and assistance. However, after running on 64 cores for 6 hours, I encountered a “bond atom missing” error. system_npt.data
Since this instability is rare, I would guess that it is not due to a bug in ‘fix bond/react’ nor any major mistake in your input. Instead, you can tune various parameters for stability; I would start with the ‘stabilize_steps’ keyword. Some reactions require stability for longer than the default 60 timesteps (could try 100 and go from there).
Sir, thank you very much for your suggestion. One method I have currently considered is to prevent the missing of atoms in C-H and O-H bonds via the command: ***fix xx all shake 0.0001 20 0 b xx xx*** . The second method is the one you proposed.
However, when I inserted stabilize_steps 100 after the prob 0.0002 parameter, as shown below: fix rxns all bond/react stabilization yes statted_grp .3 &
** react rxn1_stp1 all 1 2.5 3.5 mol1 mol2 rxn1_stp1.map prob 0.0002 stabilize_steps 100 12345 &**
** react rxn1_stp2 all 1 2.0 3.5 mol3 mol4 rxn1_stp2.map prob 0.0001 stabilize_steps 100 12345 &**
** react rxn1_stp3 all 1 2.0 3.5 mol5 mol6 rxn1_stp3.map prob 1 stabilize_steps 100 12345**
running the script triggered the following error:
ERROR: Expected integer parameter instead of ‘stabilize_steps’ in input script or data file (src/REACTION/fix_bond_react.cpp:349)