Hi Lammps users,
I’m trying to Simulate the reaction between K₂O and SiO₂ at 550°C.Compile the Born-Mayer-Huggins (BMH) potential function based on the article https://www.sciencedirect.com/science/article/pii/S0022309324000243?via%3Dihub, but encounter issues with atom loss and excessive pressure. Seeking assistance.
My data file and the input file are as follows,
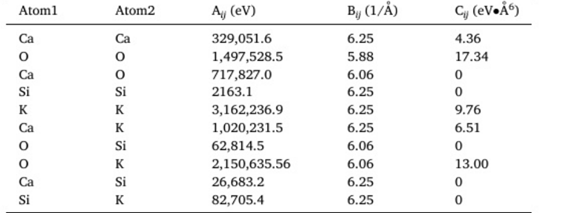

Input
units metal
boundary p p p
atom_style charge
dimension 3
timestep 0.001
neighbor 2.0 bin
neigh_modify delay 0 every 1 check yes
variable Time equal step*dt
#------------ system defination-------
read_data model.data
set type 1 charge 4.0
set type 2 charge -2.0
set type 3 charge 1.0
#----------------- setup-------
pair_style buck/coul/long 10.0
pair_coeff 1 1 2163.10000 6.25 0.0 # Si-Si
pair_coeff 2 1 62814.5000 6.06 0.0 # O-Si
pair_coeff 2 2 1497528.50 5.88 17.34 # O-O
pair_coeff 2 3 2150635.56 6.06 13.0 # O-K
pair_coeff 3 3 3162236.90 6.25 9.76 # K-K
pair_coeff 1 3 82705.4000 6.25 0.0 # Si-K
kspace_style ewald 1.0e-4
------------------------ Enegy minimization ---------------------------------------------------------------------------------------#
thermo 100
thermo_style custom step temp etotal ke pe press
dump All all custom 100 All_atom_823K.lammpstrj id type element x y z
dump_modify All element Si O K
dump_modify All sort id
min_style cg
minimize 1.0e-4 1.0e-6 100 1000
velocity all create 300 825333 mom yes rot yes dist gaussian
fix 1 all nvt temp 300 823 0.1
run 800000
Data file
HEADER
9516 atoms
3 atom types
0.000000000000 56.500000000000 xlo xhi
0.000000000000 56.560000000000 ylo yhi
0.000000000000 65.990000000000 zlo zhi
Masses
1 28.08500000 # Si
2 15.99900000 # O
3 39.09830000 # K
Atoms # charge
1 1 4.000000 5.591000000000 52.428000000000 3.223000000000
2 2 0.000000 54.206000000000 1.874000000000 2.132000000000
3 2 0.000000 0.774000000000 18.322000000000 11.283000000000
4 2 0.000000 20.249000000000 12.136000000000 4.812000000000
5 2 0.000000 13.112000000000 6.979000000000 15.471000000000
6 1 4.000000 47.935000000000 1.309000000000 9.889000000000
7 2 0.000000 8.386000000000 4.478000000000 13.859000000000
8 2 0.000000 14.469000000000 37.099000000000 5.509000000000
9 2 0.000000 16.310000000000 18.894000000000 11.248000000000
10 1 4.000000 41.153000000000 24.504000000000 7.955000000000
11 2 0.000000 52.886000000000 9.634000000000 5.602000000000
12 2 0.000000 1.640000000000 11.281000000000 11.979000000000
13 2 0.000000 23.265000000000 12.014000000000 10.270000000000
14 1 4.000000 9.151000000000 51.948000000000 17.128000000000
15 2 0.000000 49.917000000000 6.020000000000 19.471000000000
16 2 0.000000 54.204000000000 0.854000000000 5.557000000000
…………