Hi,
Thank you very much for your reply.
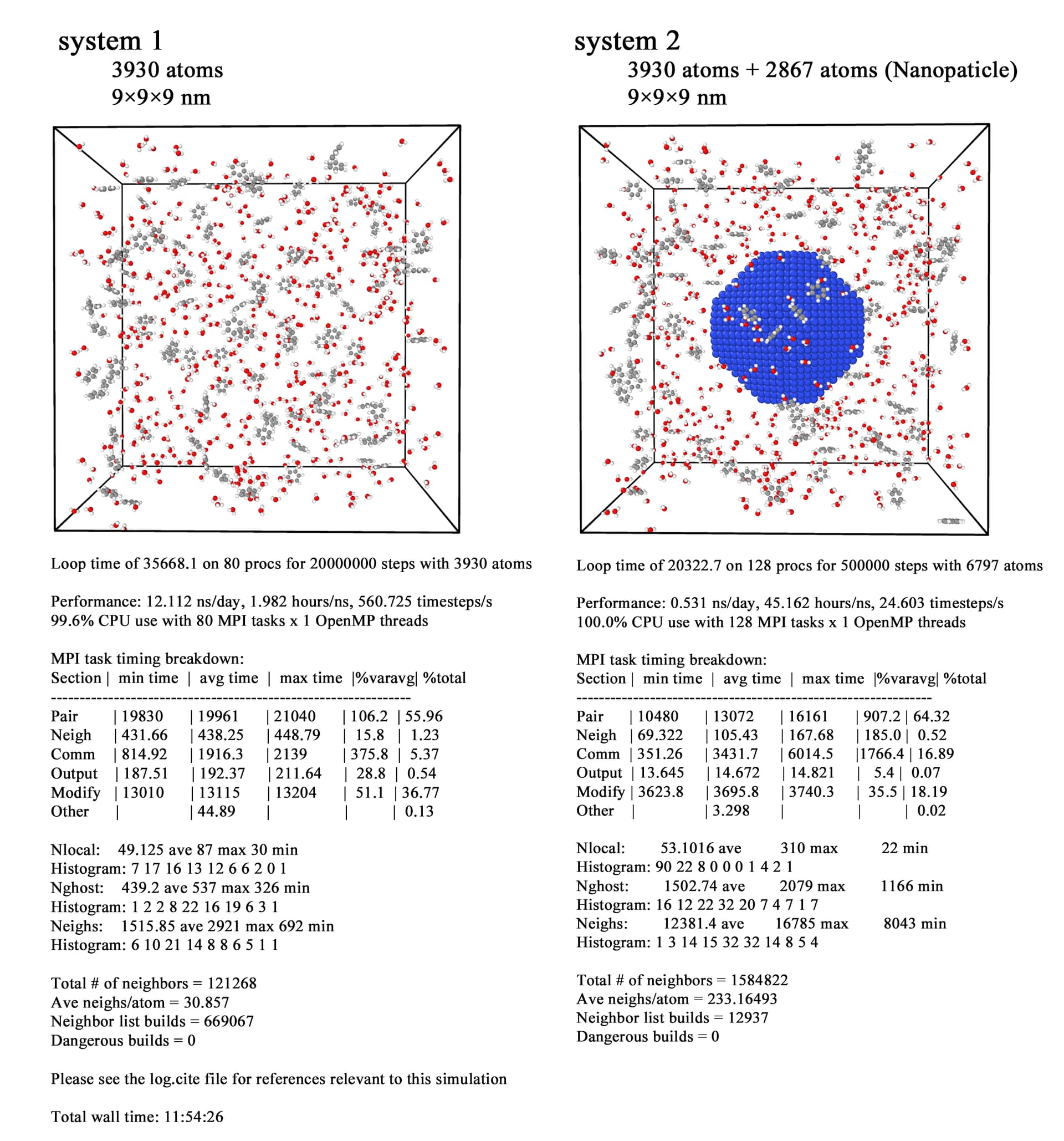
The details of the simulation systems are shown in the following figure.
The input file for system 1:
units real
boundary p p p
atom_style charge
read_data C10W600.data
pair_style reax/c lmp_control safezone 16 mincap 10000
pair_coeff * * CHONSFPtCl.ff C H O N S F Pt Cl X
neighbor 2 bin
neigh_modify every 10 delay 0 check yes
velocity all create 300 4928459 rot yes dist gaussian
timestep 0.25
fix 1 all qeq/reax 1 0.0 10.0 1e-6 reax/c
fix 2 all nvt temp 300.0 300.0 25.0
thermo_style custom step temp press density
thermo 100
run 19400
unfix 2
reset_timestep 0
fix 4 all nvt temp 300.0 2000.0 25.0
fix 6 all reax/c/species 1 1 1000 C10W600-2000K.out element C H O N S F Pt Cl X
fix 7 all reax/c/bonds 10000 C10W600-2000K-bonds.txt
dump TRAJ all custom 100 C10W600-2000K.xyz id type element x y z
dump_modify TRAJ element C H O N S F Pt Cl X
restart 100000 Restart.restart
run 500000
The input file for system 2:
units real
boundary p p p
atom_style charge
read_data C10W600-4.data
pair_style reax/c lmp_control safezone 16 mincap 10000
pair_coeff * * CHONSFPtCl.ff C H O N S F Pt Cl X
neighbor 2 bin
neigh_modify every 10 delay 0 check yes
velocity all create 300 4928459 rot yes dist gaussian
timestep 0.25
group NPs type 9
group ben type 1 2 3
fix 1 all qeq/reax 1 0.0 10.0 1e-6 reax/c
fix 2 all nvt temp 300.0 300.0 25.0
fix 3 NPs momentum 100 linear 1 1 1 angular
thermo_style custom step temp press density
thermo 100
comm_style tiled
fix fixbalance all balance 500 1 rcb weight group 2 NPs 30 ben 1.0
run 19400
unfix 2
reset_timestep 0
fix 4 all nvt temp 300.0 2000.0 25.0
fix 6 all reax/c/species 1 1 1000 C10W600-4-2000K.out element C H O N S F Pt Cl X
fix 7 all reax/c/bonds 10000 C10W600-4-2000K-bonds.txt
dump TRAJ all custom 100 C10W600-4-2000K.xyz id type element x y z
dump_modify TRAJ element C H O N S F Pt Cl X
restart 100000 Restart.restart
run 500000
**lmp_control file **
simulation_name Blends_LowT
tabulate_long_range 10000
energy_update_freq 1
nbrhood_cutoff 4.5
hbond_cutoff 6.0
thb_cutoff 0.001
bond_graph_cutoff 0.3
write_freq 10000
traj_title Blends_LowT
atom_info 1
atom_forces 0
atom_velocities 0
bond_info 0
angle_info 0
Many thanks for your help.