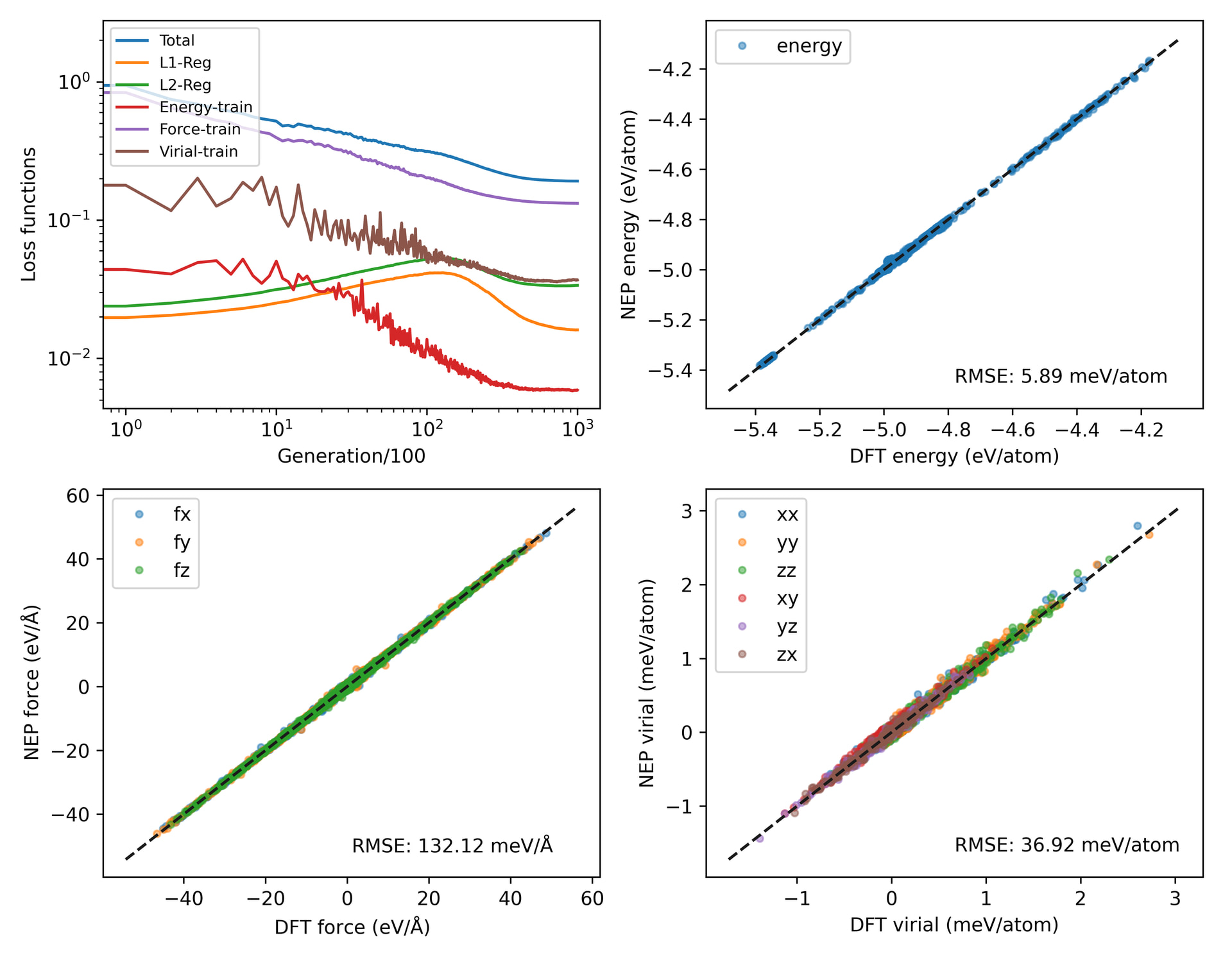
Below are the details of my potential function training:
My goal is to train a potential function for the solid-liquid interface. The dataset includes three types: solid structure, liquid structure, and solid-liquid interface structure. The initial dataset is obtained through perturbation of the structures, as well as the structures obtained by running nep89 in the npt ensemble for 1 ps. After obtaining a good initial potential function from the above datasets, I use this potential function to run active learning to obtain a 10 ps structure. Then, I merge the above data and run a new potential function. Through this active learning process from 1 ps to 10 ps - 100 ps - 1 ns - 10 ns, I have trained and iterated the existing potential function. The 10 ns active learning run.in, the 10 ns test structure loss graph, and the latest potential function loss graph are shown as follows. However, the active learning from 1 ps to 10 ps is in the npt ensemble, but at 100 ps, the structure obtained by running in the npt ensemble is abnormal. But it was found that using the nvt ensemble resulted in normal structures. Therefore, from 100 ps to 1 ns to 10 ns, the nvt ensemble was used. I am puzzled as to why the active learning in the npt ensemble cannot obtain reasonable structures while the nvt ensemble can. I do not know if the final potential function is sufficiently reasonable? I sincerely request experts to help analyze.
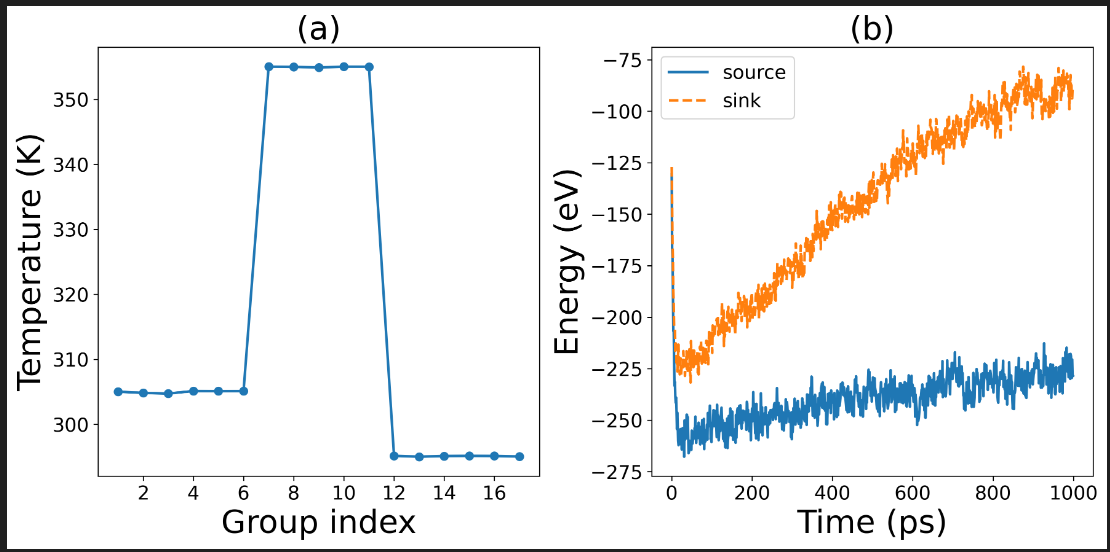
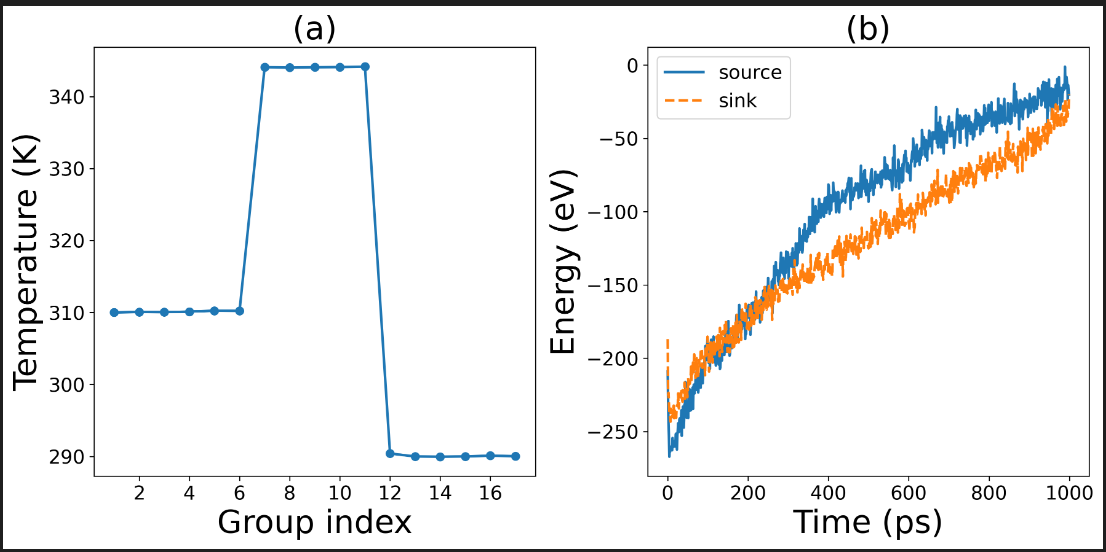
Next, I used my latest potential function to perform the nemd calculation. However, this process encountered an anomaly. The nemd calculation was able to complete, as shown in the following figure. It was attempted twice. The first time, the heat source and heat sink temperatures were 295K and 305K respectively, and it ran for 1ns; the second time, the heat source and heat sink temperatures were 290K and 310K respectively, and it ran for 5ns. Both times, there were abnormal temperature distributions in the middle part of the liquid structure and abnormal energy diagrams of the heat source and heat sink. The cause of this anomaly is still unknown. We sincerely request experts to help analyze it.
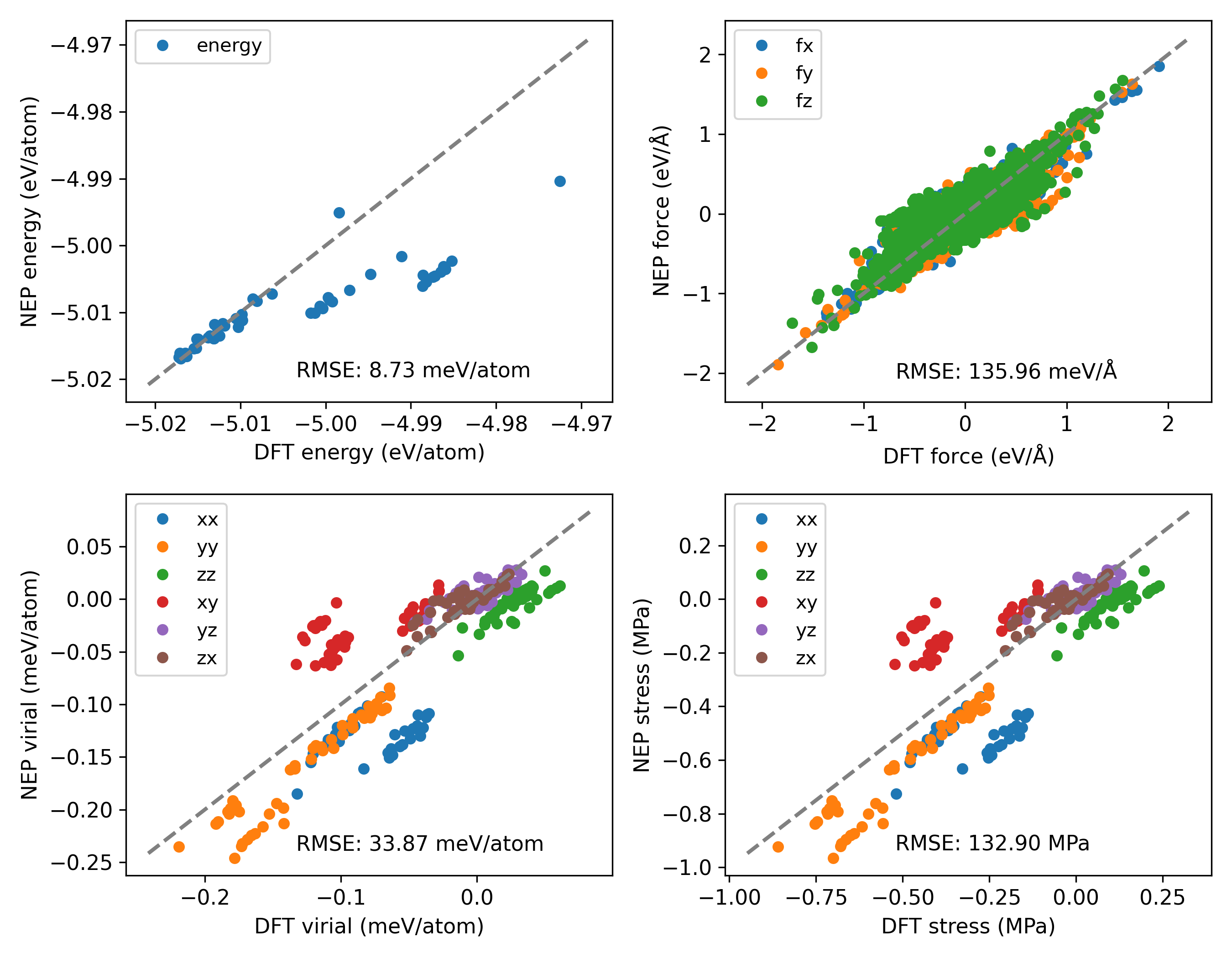
Your parity plots for the test set looks odd to me. What are the outlier structures that fall below the parity line in the energy (upper left panel of prediction_SWS_test…)? Are they different to the ones that are on the parity line?
In your training set (second figure) you also seem to have a lot of structures with very large forces (>10 eV/Å). Having some structures with large forces is good for model stability, but having structures with too large forces can make the model worse. Try filtering out structures with forces >10 eV/Å and see if that helps with stability in the npt ensemble.
Can you share your nep.in file? When you perform active learning, how many structures do you select and how do you choose them?
Unfortunately I’m not too familiar with NEMD, I’ll leave that to someone else.
This is the explanation for your first question. All the test structures are of the same structure. There is indeed a part that is slightly lower than the matching line, possibly due to the influence of temperature changes. Because these structures include those with temperatures of 280K, 290K, 300K, 310K, and 320K. However, I believe the energy range is already very small (-5.02 to -4.97 eV/atom), so this error is within my acceptable range. I think it won’t have a significant impact. This is my humble opinion, but as of now, I don’t know if it has a significant effect.
For the second question, my structure includes perturbation and active learning parts. Maybe the structures with large forces should mainly come from the perturbation. Previously, some teachers told me that a force of 50 eV/Å is acceptable, so I retained these structures. To eliminate the influence of excessive forces, I plan to try retaining the 10 V/Å structures and retrain the potential function. I would like to ask, are you busy? Based on the screening, can you share it?
Here is my nep.in. My initial training structures are approximately 300 to 400 in number. Later, I mainly used active learning to run interface structures. In each round of active learning, 50 new interface structures were obtained. A total of 4 rounds of active learning were conducted. You can share your training experience, please?
Aha I see, that makes sense since the absolute energy range is quite small. Are you training the model for 300K? In some cases it can be useful to do active learning over a broader range of temperatures and pressures (perhaps 200K to 500K and 0 GPa to 10 GPa, depending on what makes sense for your system) to increase the model stability. Sometimes this requires some care when selecting structures from the active learning run, as some of them might have very large forces.
Yes, you could try removing the structures with the large forces. What do you mean with “Based on the screening, can you share it?”?
50 structures per active learning round sounds reasonable. How do you select what 50 structures to choose? Is the initial set of structures (the 300-400 structures) all from perturbation, or did you obtain some of them in another way? Do you notice an improvement in the model performance and stability for each round of active learning?
Hello, I just saw this post. I think you have done the active learning process quite well. And I am not concerned by the realtive large forces in some of the structures. You can keep them. What you need to do are two folds:
Continue to do the 10-ns MD run and test, to see if the test accruacy improves compared to the previous try. They should improve and finally converge.
After the test accuracy for 10-ns MD run is very good and converged, you should solve the NPT instability problem, beforing moving to the NEMD simulations. I guess you might have not correctly set up the NEMD simulations, but we will check that later after the NEP model is finalized.
To check if the test accuracy improves, I would like to see the parity plots for the test set using the NEP models trained before and after adding this test set.
hello,my initial structure was obtained using perturbation and MD, and the stability is enhanced with each iteration, as can be seen from the increased number of test steps that can be run after each iteration.
Thank you for your reply, Professor. I have made some attempts and successfully obtained reasonable structures by running 1ns of active learning in the NPT ensemble with the current potential function. I believe the current potential function has a certain degree of stability, though there might be some minor details that need refinement. I will continue testing the structure for 10ns and retrain the potential function for another generation with this test set. Additionally, I have made some improvements to the NEMD instructions as follows: changing the timestep to 0.5, increasing the relaxation time to 1ns, and running NEMD for 5ns. This task is currently still under computation.
With H atoms, you indeed need a timestep of 0.5 fs. As for NEMD simulations, you need to know that the grouping method is static, meaning that a group for the liquid might loose its spatial locality during the MD simulations. The solid part is ok. Keep this in mind and I am looking forward to your new results.
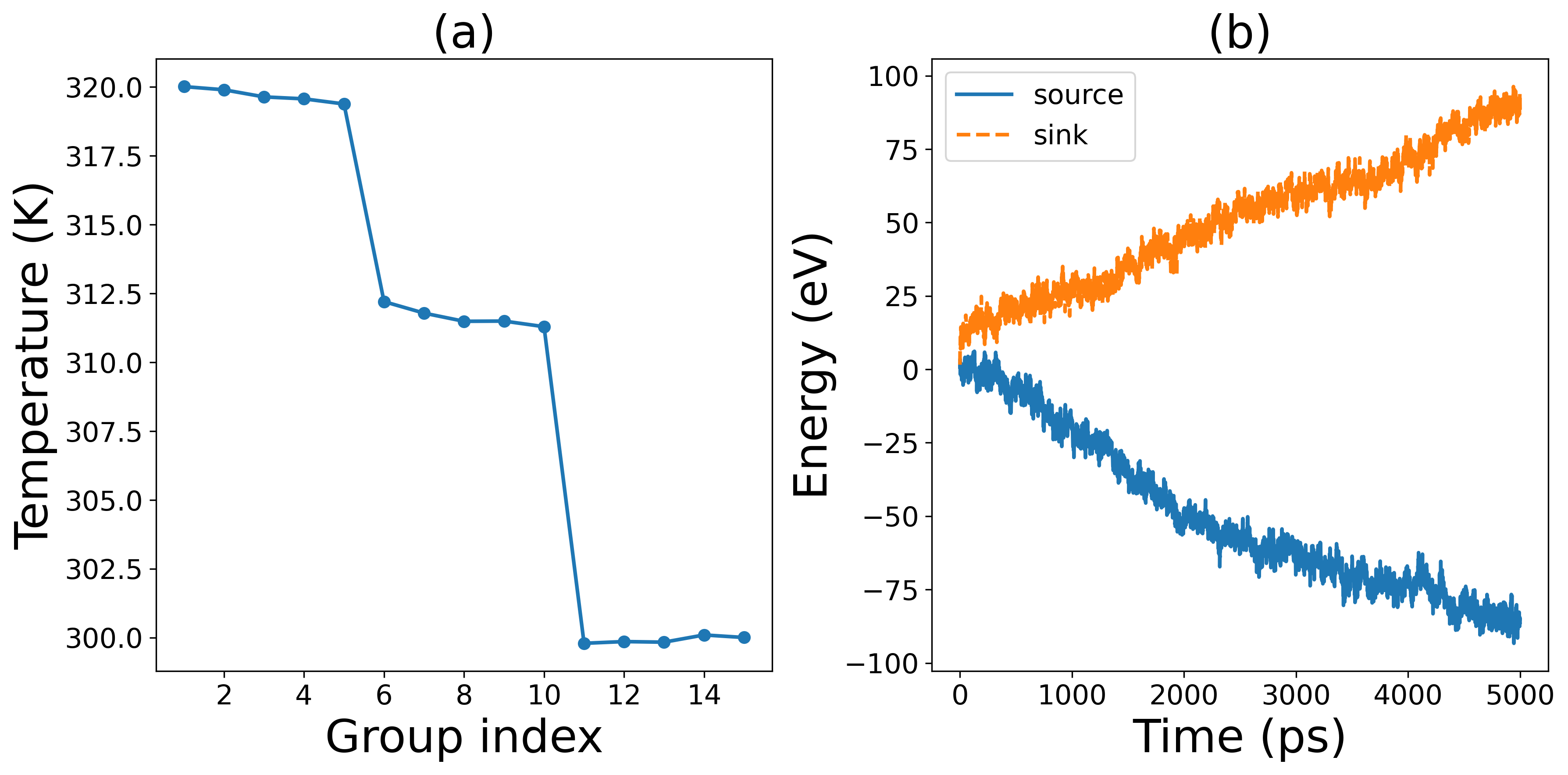
Hello professors and experts, I have improved the NEMD input parameters by using timestep=0.5, a relaxation time of 1 ns, and a calculation duration of 5 ns. The resulting temperature and energy graphs show some improvements. The final interfacial thermal conductivity was determined to be 4 MW/m^2*K. Although there are few references using GPUMD to calculate this interface property, by comparing with other methods and materials, I estimate that my calculated value is about an order of magnitude lower than the ideal value. I checked the calculation formulas and found no errors. Could it be that my potential function is not accurate enough? What should I do?
Hello professor, I just searched for the experimental measurement values related to this study. I found that the team led by Professor Jiang Puqing from Huazhong University of Science and Technology obtained a measurement value of 5.7 MW/m^2*K. Their team also confirmed the accuracy of these measurements. This suggests that my calculated values are somewhat reasonable. However, I think more verification is needed. What kind of analysis do you think I should conduct? Should I perform some spectral heat flux analysis?
The NEMD results look better now but it is difficult to follow your presentation. We actually do no know what are improved and solved.
I strongly suggest you solve the problem one by one. I would like to go back to the the active learning process and see how the results are improved little by little. Both NVT and NPT.
Okay, I understand. Actually, I’m currently running the NPT test set as well. This NEMD calculation uses the same potential function as mentioned above, but I’ve made improvements in the relaxation and calculation steps. I’m still refining the potential function through active learning, and I’ll share my improvement results later.