I want to simulate a simple crosslinking reaction with fix bond/react command in LAMMPS. I’ve checked the models before the crosslinking reaction with write_data command, but I don’t know why the model after crosslinking reaction cannot be opened with OVITO.
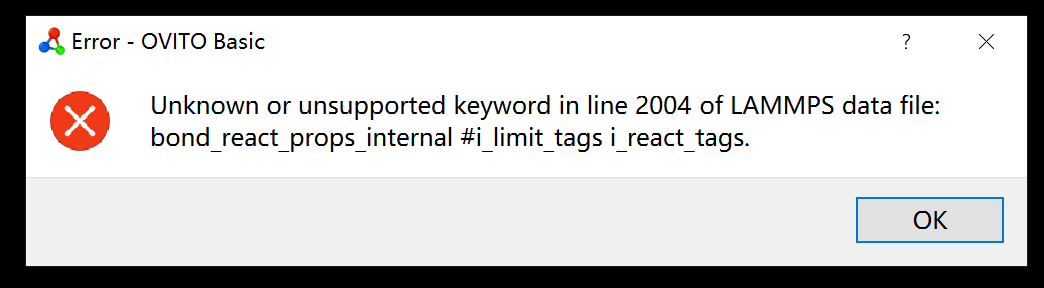
Following is my in.file.
units real
boundary p p p
atom_style full
bond_style harmonic
angle_style harmonic
dihedral_style nharmonic
pair_style lj/gromacs 12 15
read_data Polymer.data extra/bond/per/atom 1 extra/angle/per/atom 1 extra/dihedral/per/atom 1
mass * 74
bond_coeff * 115.4086 2.801
angle_coeff * 64.62431 111.623
dihedral_coeff * 4 3.280141429 -0.59019769 1.991530534 3.31026047
variable T equal 400 # TARGETT
pair_coeff * * 0.36 4.5
neighbor 2 bin
neigh_modify every 1 delay 5
timestep 5
thermo 1000
thermo_style custom step etotal ke temp pe ebond eangle evdwl press vol lx ly lz density
velocity all create ${T} 7856347
minimize 1.0e-10 1.0e-10 10000 15000
reset_timestep 0
fix 11 all nvt temp $T $T 500 drag 2 # iso 1000 1000 10
run 10000
unfix 11
####### crosslinking ########
molecule mol1 pre-molecule.data
molecule mol2 post-molecule.data
reset_timestep 0
fix rxns all bond/react stabilization yes statted_grp .03 reset_mol_ids yes &
react myrxn1 all 100 0 7 mol1 mol2 map-molecule.data
fix 2 statted_grp_REACT nvt temp $T $T 500
run 2000
write_data m.data
Following is my pre-molecule.data:
read using pysimm.system.read_mol2 Bonding_Atoms 4 14
8 atoms
6 bonds
4 angles
2 dihedrals
Types
1 1
2 1
3 1
4 2
5 3
6 1
7 1
8 1
Charges
1 0
2 0
3 0
4 0
5 0
6 0
7 0
8 0
Coords
1 0 0 0
2 3.66 0 0
3 7.32 0 0
4 10.98 0 0
5 14.64 0 0
6 18.3 0 0
7 21.96 0 0
8 25.62 0 0
Bonds
1 1 1 2
2 1 2 3
3 1 3 4
4 1 5 6
5 1 6 7
6 1 7 8
Angles
1 1 1 2 3
2 1 2 3 4
3 1 5 6 7
4 1 6 7 8
Dihedrals
1 1 1 2 3 4
2 1 5 6 7 8
Following is my post-molecule.data:
read using pysimm.system.read_mol2 Bonding_Atoms 4 14
8 atoms
7 bonds
6 angles
5 dihedrals
Types
1 1
2 1
3 1
4 2
5 3
6 1
7 1
8 1
Charges
1 0
2 0
3 0
4 0
5 0
6 0
7 0
8 0
Coords
1 0 0 0
2 3.66 0 0
3 7.32 0 0
4 10.98 0 0
5 14.64 0 0
6 18.3 0 0
7 21.96 0 0
8 25.62 0 0
Bonds
1 1 1 2
2 1 2 3
3 1 3 4
4 1 4 5
5 1 5 6
6 1 6 7
7 1 7 8
Angles
1 1 1 2 3
2 1 2 3 4
3 1 3 4 5
4 1 4 5 6
5 1 5 6 7
6 1 6 7 8
Dihedrals
1 1 1 2 3 4
2 1 2 3 4 5
3 1 3 4 5 6
4 1 4 5 6 7
5 1 5 6 7 8
Following is my map-molecule.data:
8 equivalences
2 edgeIDs
BondingIDs
4
5
EdgeIDs
1
8
Equivalences
1 1
2 2
3 3
4 4
5 5
6 6
7 7
8 8
I really hope someone could help me!