Dear developer,
When I try to run the example, Si, in Amset, I found that setting the doping option to zero will fail the calculation.
I think a material which without doping should works.
I wonder how the “doping” works in Amset?
From the document, “Controls which doping levels (carrier concentrations) to calculate”.
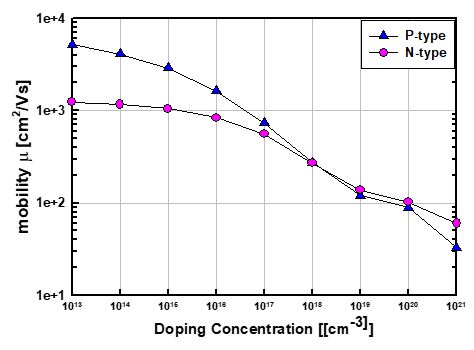
Is this “doping” in Amset a real doping or carrier, such as electron or hole?
Does the mobility calculated from Amset from n- or p-type doping means the mobility of electron or hole?
Best,
John Chen