My question has to do with the total energy conservation of the NVE integrator in LAMMPS with a dihedral potential applied to a short toy polymer. I am using the Aug 2023 distribution of LAMMPS.
Here is the system I am considering:
There are four atoms connected in a chain with harmonic bonds (1 bonds 2, 2 bonds 3, 3 bonds 4). These atoms interact with a LJ potential (FENE bonds are on such that only 1-2 pairwise interactions are excluded). Additionally, there is a dihedral potential applied to this polymer chain. The system is initialized with a velocity corresponding to 300 Kelvin.
I am observing that when I simulate the system with a dihedral potential on (I tested a quadratic dihedral potential and a harmonic dihedral potential), total energy is not conserved (it increases with time). I tested a variety of systems with different boundary conditions and various potentials turned on and off. Here are those systems:
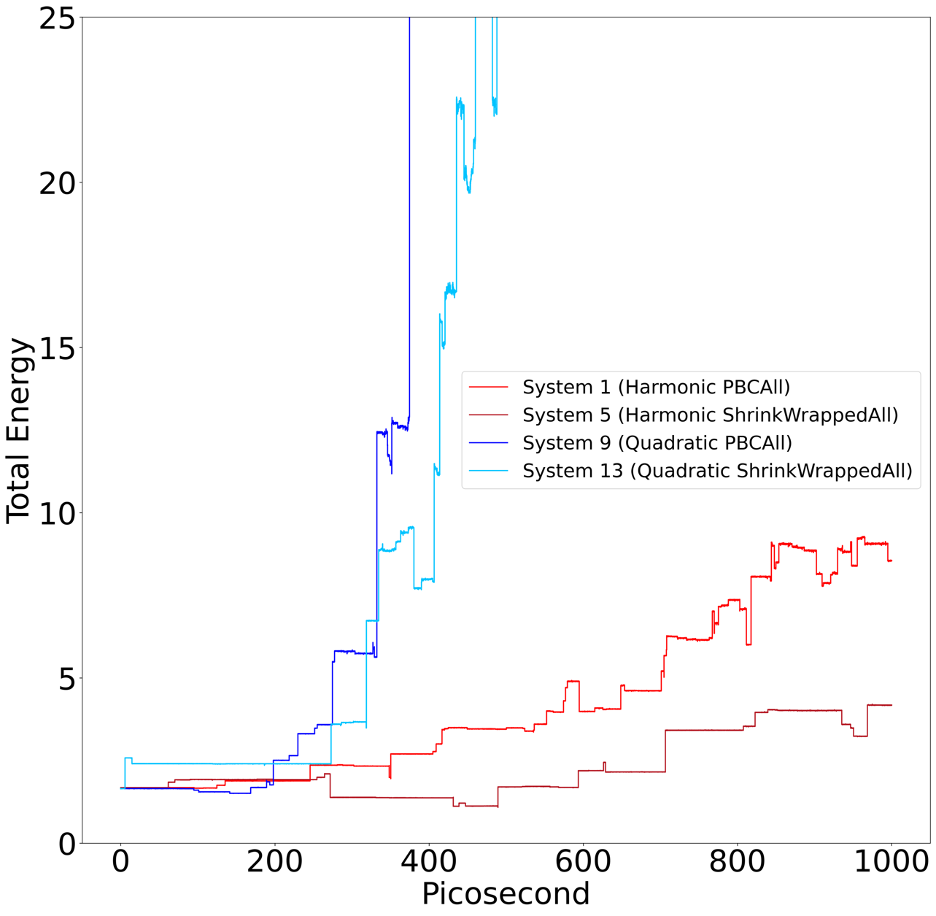
System 1 – Harmonic PBCAll:
Harmonic dihedral potential, LJ potential, harmonic bonds, periodic boundary conditions. Energy is not conserved.
System 2 – Harmonic PBCBondedOnly:
Harmonic dihedral potential, harmonic bonds, periodic boundary conditions. Energy is not conserved.
System 3 – Harmonic PBCDihedralOnly:
Harmonic dihedral potential only, periodic boundary conditions. Energy is not conserved.
System 4 – Harmonic PBCNoDihedral:
LJ potential, harmonic bonds, periodic boundary conditions. Energy is conserved.
System 5 – Harmonic ShrinkWrappedAll:
Harmonic dihedral potential, LJ potential, harmonic bonds, shrink wrapped boundary conditions. Energy is not conserved.
System 6 – Harmonic ShrinkWrappedBondedOnly:
Harmonic dihedral potential, harmonic bonds, shrink wrapped boundary conditions. Energy is not conserved.
System 7 – Harmonic ShrinkWrappedDihedralOnly:
Harmonic dihedral potential only, shrink wrapped boundary conditions. Energy is conserved.
System 8 – Harmonic ShrinkWrappedNoDihedral:
LJ potential, harmonic bonds, shrink wrapped boundary conditions. Energy is conserved.
System 9 – Quadratic PBCAll:
Quadratic dihedral potential, LJ potential, harmonic bonds, periodic boundary conditions. Energy is not conserved.
System 10 – Quadratic PBCBondedOnly:
Quadratic dihedral potential, harmonic bonds, periodic boundary conditions. Energy is not conserved.
System 11 – Quadratic PBCDihedralOnly:
Quadratic dihedral potential only, periodic boundary conditions. Energy is not conserved.
System 12 – Quadratic PBCNoDihedral:
LJ potential, harmonic bonds, periodic boundary conditions. Energy is conserved. This is a duplicate of system 4.
System 13 – Quadratic ShrinkWrappedAll:
Quadratic dihedral potential, LJ potential, harmonic bonds, shrink wrapped boundary conditions. Energy is not conserved.
System 14 – Quadratic ShrinkWrappedBondedOnly:
Quadratic dihedral potential, harmonic bonds, shrink wrapped boundary conditions. Energy is not conserved.
System 15 – Quadratic ShrinkWrappedDihedralOnly:
Quadratic dihedral potential only, shrink wrapped boundary conditions. Energy is conserved.
System 16 – Quadratic ShrinkWrappedNoDihedral:
LJ potential, harmonic bonds, shrink wrapped boundary conditions. Energy is conserved. This is a duplicate of system 8.
Each of these systems are in the dropbox link I have included here with inputs, configuration files, potential files, trajectories, and log files:
The files are labelled according to the title after System # – FolderTitle.
My original expectation before running these simulations was that if all the energy functions were well-posed and their forces were derived properly, energy should be conserved (given a sufficiently small time step and relatively small forces). Because I am not seeing that, I have a few questions about the behavior I am observing:
- Why does the NVE integrator not conserve energy when a dihedral potential is applied (except in the case of a dihedral potential applied to a polymer chain with shrink wrapped boundary conditions and no other potentials)?
- Why is there a significant difference in the rate of total energy increase between the same system simulated with PBCs versus shrink wrapped boundary conditions? The boundary conditions for the PBCs should be a box that is large enough such that periodic images do not interact (260^3 cubic angstroms versus a chain that at full stretch is ~12 angstroms).
To motivate question 2, I have included a graph of the total energy in the systems 1, 5, 9, and 13. If there are any additional questions about my simulations, please let me know. Any insight would be extremely helpful.

As a side note: I ran system 1 at a timestep of 0.1 femtoseconds and the behavior was similar to the 10 femtosecond timestep I was using in the prior simulations. Those files can be found here: