Hi everyone, recently I encountered the following two problems while doing lammps simulation.
1.Recently, I was doing the bending simulation of Si beam structure, and I found that my model did not break as expected under the action of constant speed loading, but bent to a certain extent and then produced rebound (the speed I set was constant). However, when I increased the size of the simulation model, certain fracture would occur, but as the simulation continued, When the model is bent to a certain extent, it will bounce back. I don’t know why this is, and I kindly ask you to help me correct it.
2.I want to use a small model because I want to simplify the calculation and thus reduce my simulation time. If the conclusion of the above question is that I must increase the simulation size, I would like to ask whether I am more efficient at the simulation device level by using GPU acceleration or by purchasing a unified memory machine (or something else).
Attached below are my input files, log files, and generated graphics files (i put it in the link,it about 26.8MB).
In response to your point 1: Your description is far too vague to make any meaningful statement, but also, this is not really a LAMMPS questions, but something you need to discuss with your adviser or supervisor or experienced colleagues, i.e. people that know and care about your research.
In response to your point 2: The choice of system size should not be determined by your available compute resources, but by the science of your problem. It most certainly cannot be determined by random people on the internet that neither know you nor your research.
You are using a KIM model and - to the best of my knowledge - those are not GPU accelerated at all. You may have to ask in the OpenKIM forum elsewhere here on MatSci.org to be certain.
Thank you for your reply. I think I did not describe the first question clearly.
In my mind, if I set a constant speed for a group of atoms, they will keep moving, but this is not the case in my model, first I set a constant speed for the model:
The nvt ensemble is used globally, and the constant temperature simulation is carried out:
Then a simulation with a time step of 0.001 and an image output every 1000 steps was performed for a total of 50 picoseconds. (The global unit is metal)
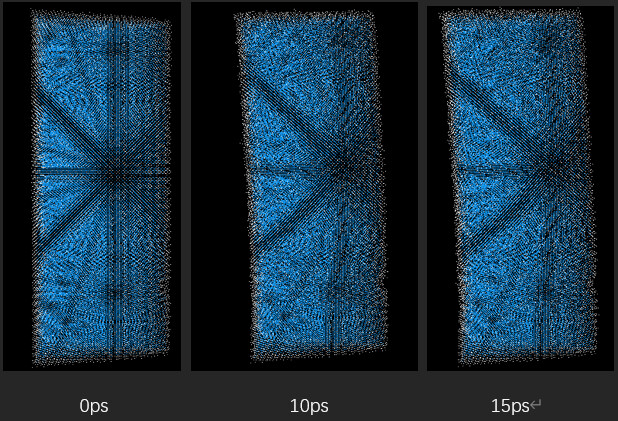
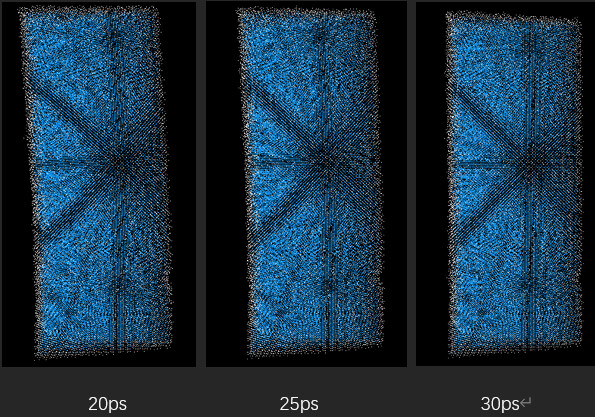
But when I looked at the simulation results (using ovito software), when the model moved to a certain point, it seemed to lose speed and rebound.:Here are several result graphs I generated, the time is marked below:
The image shows that when the time reaches about 15 picoseconds, the motion stops. As the simulation continues, the model bounces back. I don’t quite understand this display result. I am wondering whether it is the setting of the environment during my simulation or the default setting of lammps during simulation.
According to Newton’s laws of motion, this will only happen when the atoms experience no forces.
Your setup, however, is more like knocking on a tuning fork. But as I wrote before, this is not a LAMMPS issue but a question of the science your are trying to represent and thus it is something to discuss with those that have a vested interest in it.