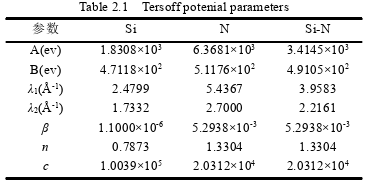
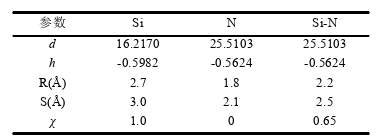
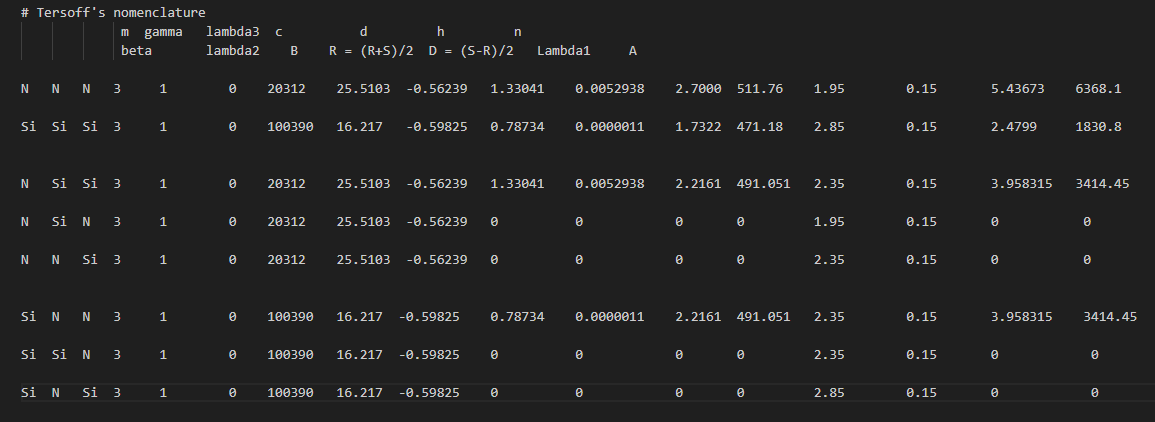
Recently, I am simulating the molecular dynamics of silicon nitride, using tersoff potential function, and writing tersoff potential function according to the parameters given in the paper, but I can not get the desired effect. The following is attached the potential function written by me and the data given in the paper. Please help me to look at the problems in the documents I have written. I would be very grateful if you could provide me with the tersoff file of Si3N4!!
Maybe a stupid question, but are you sure you used the correct units for the sake of the “units” command you are using in the input script? Maybe you have to convert units.
Can you share your input file? (it may be easier to help you if we can see it).
The many values that you are setting to 0.0 are highly suspicious.
The best way to go about learning how to convert parameters from a publication to LAMMPS style potential files is to practice with some publications for which equivalent files already exist in the LAMMPS potentials folder. Those usually contain the reference where the data is taken from. This way, you can easily check whether your input matches when is in the provided files. If not, you can then more easily reverse engineer how the conversion should have been done. This should then inform how to convert any equivalent file for the same style of potential.
I am very glad to receive your reply. For the Settings of some 0.0 parameters in the potential function, I set them according to the definition of two-body potential and three-body potential of tersoff potential function in lammps manual, and the fitting of potential function is based on the reference of tersoff potential function of SiC provided by lammps software. I still don’t know where the problem is. There are many disputes about the most doubtful parameter n, but I don’t know how to modify it. Please help me have a look again or help me find out if there is a correct tersoff potential function of Si3N4 that has been fitted. Thank you very much!
I have given you advice how you can teach yourself how to do this correctly. You say that you followed the instructions, but I suspect that that is not correct. My suggestions help to resolve this. However, you seem to prefer to ignore it. That is fine by me, but don’t expect me to do what is essentially your job.