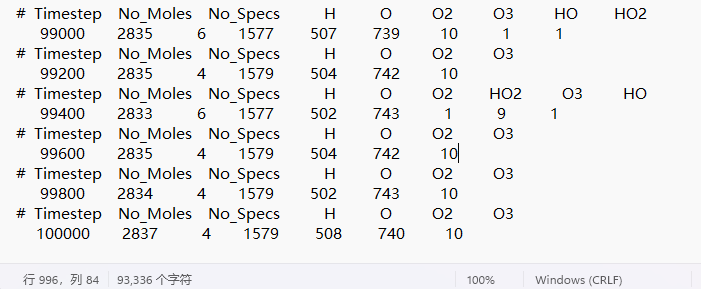
I want to study the reaction between ozone and water. So I did 20 ns of reaxff force field, 0.2 fs in length, 100000 steps in total. And using fix reaxff/species, I get H and O, and I get oxygen, but for some information, I should get oxygen, hydrogen peroxide, or hydroxyl radical. May I ask why I made a mistake.
It’s impossible to provide any advice without sharing your simulation setup. Please read the forum guidelines before replying.



You have clear inconsistencies between your data file and your pair_coeff.
@lihao1 Seems that you missed that important part:
Sharing snapshots is very bad practice on here.
Hard to say. Was your ReaxFF potential file parameterized for the chemical reactions you want to study? And the conditions you are using?
I’m sorry, I can’t send the original file because I’m a new user. The system does not allow me to send. But since I’m using the REAXFF force field, I shouldn’t need to set up pair coeff in the data file. I have set before, but will directly error.
I‘m sorry, as a new user, I want to know what is reaxff’s potential file parameterizes my chemical reaction.Could you help me to explain? I think my condition has been written in the in file, I do not know if you also refer to any other conditions.Thank you.Here is my in file.

Thats not what I asked.
You still have a pair_coeff command, which is wrong.
This discussion is circular, and you clearly refuse to read the guidelines of the forum. I am out.
1 Like
To figure out which ReaxFF potential file is suitable for your system is your job not mine. Every potential file has an associated publication and that publication describes what the parameter set what trained for. The file name in your screenshot (very, very bad idea to use those, BTW) suggests that it may be the force field for combustion reactions between CO, H2O, CO2, HCHO, HCOOH, and so on. That would most certainly not be suitable to represent bulk water at 300K. In fact, a good benchmark for the force field would be to run a simulation of pure bulk water and compare the structure (e.g. g(r) for O-O, O-H, and H-H) to those for well known and tested water potentials (e.g. TIP3P, SPC/E, TIP4P) and experimental data.
You are not getting the point and it also seems, that your knowledge of ReaxFF and how to use it is insufficient. So my suggestion is to study that first and do not try to run simulations with models and methods that you are lacking insight into. You are very likely to make (very) bad choices and get bogus results.
When you use the pair_coeff command, you defined * * ffield.reax H O, but in your data file, atom type 1 corresponds to oxygen (O) and atom type 2 to hydrogen (H). So, you need to reverse the order in your command to * * ffield.reax O H. By doing that, LAMMPS will simulate your system correctly!