Dear LAMMPS community Developers,
I am simulating the evaporation of water on a Cu surface using LAMMPS. Initially, I performed relaxation using the NVT ensemble and then checked the system equilibrium using NVE. The results showed a slight overall increase in system energy, but the temperature did not show a significant rise or fall. The increase in total energy was attributed to the increase in potential energy. I would like to understand what issues might have arisen in my simulation or if this slight temperature increase is normal. Can you provide insights into this matter?
Here is my in.file
in.evap (1.6 KB)
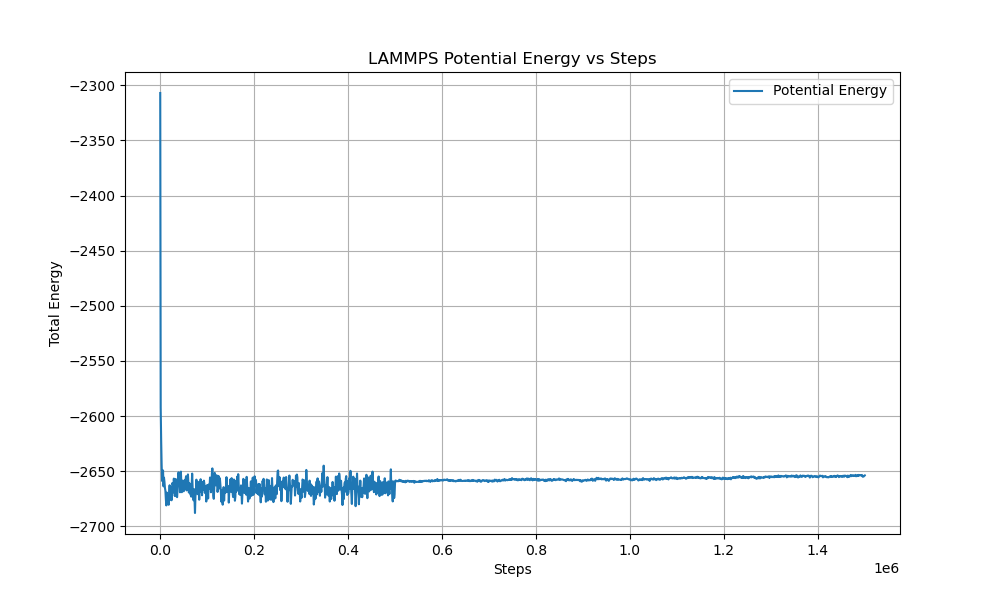
The png is the total energy, not the potential energy
This is less of a LAMMPS question but rather a question on general MD methodology and thus something that is better discussed with a local tutor or adviser or similar.
I’ve had a quick look at your input and workflow and there are some concerns
- you are using fix npt instead of fix nvt as stated in your description. this kind of inconsistencies (and also the lack of units on your graph, 0.2 steps??) are something that you have to avoid to gain people’s trust. nobody wants to figure out things only to have to ask lots of questions because of contradicting or lacking information.
- your timestep is very short (0.25fs) for your system. 1fs should be easily possible for your system. That will avoid 4x the noise from having to run 4x as many time steps for the same length of trajectory.
- you have not converted the force constant for the bond and angle potential. It has no negative effect since you are using fix shake. are you sure you will remember that you omitted the conversion in case you feel for some reason later compelled to remove fix shake? it would be cleaner to either convert everything or use bond style and angle style zero where there is no force constant to enter in the first place.
- your kspace convergence needs to be tighter for fix npt. 1.0e-4 is borderline converged even for forces, but pressure will have a large error and thus time integration with fix npt.
Some more general remarks:
- It is always difficult to pinpoint problems with complex systems. I would suggest your run the copper slab and water film separately to see if the contribution of the energy drift is similar or different between the two subsystems
- your equilibration time is rather short with 125 picoseconds.
- using fix npt for your system is not a good idea. Instead you should determine the equilibrium lattice constant for your copper potential for a bulk(!) system and then build your system based on that value and relax it with the lower part immobile (representing bulk) and fix nvt for the upper part (so that relaxation is mostly in z, plus surface reconstruction, if any). Water may be added before or after and then then be equilbrated while keeping the entire copper subsystem immobile and vice versa and only switched to the final integration settings, if those are each properly equilbrated.
- it seems you are using the TIP3P model for water. Please note that there is also a TIP3P parameter set optimized for use with long-range electrostatics as you are using.
As mentioned above, this should be discussed with your tutor, adviser, and/or senior colleagues to make sure you are making meaningful choices for your simulation. In the forum here, we may be giving you some pointers, but don’t expect much more beyond that, as this is off-topic.
Dear Axel,
I don’t know how to express my gratitude, your answer helped me a lot. With your suggestion, I have solved the problem of increasing potential energy.
Would you share with the community how you improved the energy conservation in your system? It will be a useful reference for other users.
My two cents: when the energy slowly drifts with the NVE time integrator, it is useful to examine the individual terms of the potential energy to understand which one is draining or releasing energy. The origin of energy leaking could be potential energy stored in the sample’s structure or a bad choice of parameters such as coupling constants.
1 Like
Sure, I choose the tighter kspace for 1.0e-6, and everything seem to be OK
3 Likes