Dear users, I encounter a strange phenomenon. I manage to the simulation of polymer melts at a fixed shear rate under periodic boundary conditions. However, I find that in the dump file of some steps, some atoms’ unwrapped coordinates have a dramatic change, as shown in the figure. The header line is “ITEM: ATOMS id type xu yu zu vx vy vz”.
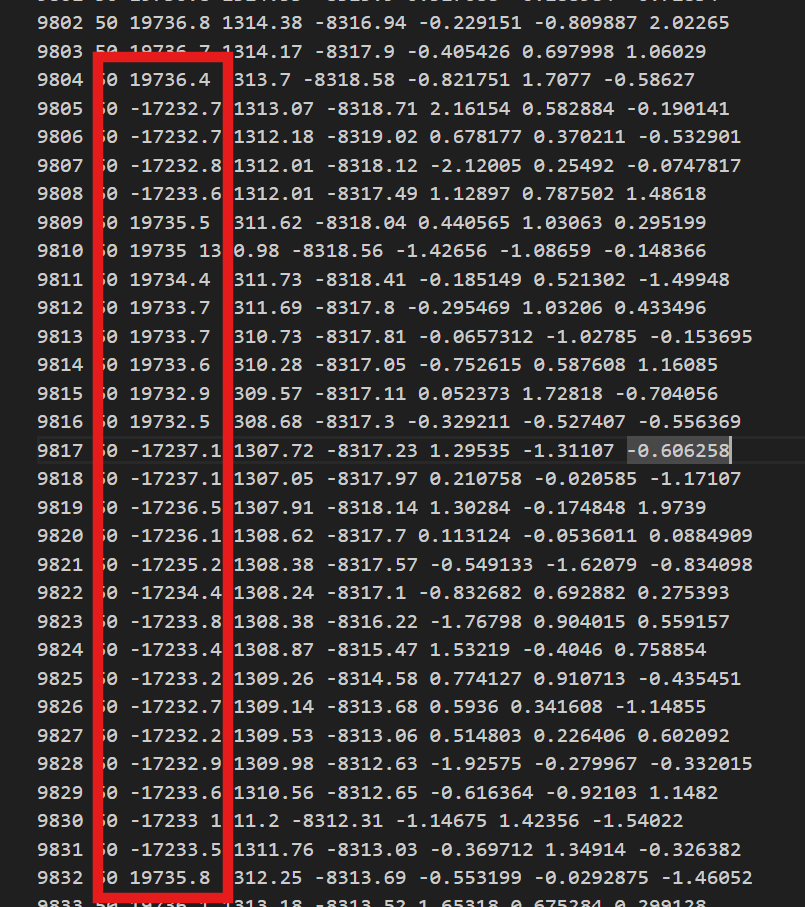
My shear and dump commands are
fix 1 all nvt/sllod temp 1 1 1
fix 2 all deform 1 xy erate 1.979797979797979e-04 units box remap v
dump 6 all custom 100000 stage6.*.${simname}.xyz id type xu yu zu vx vy vz
respectively.
Could you please tell me whether I can adjust the atomic coordinates by using periodic boundary conditions or there are other mistakes? I would very appreciate it if you could offer some guidance about this question.
Best wishes!