Hello All.
LAMMPS (23 Jun 2022)
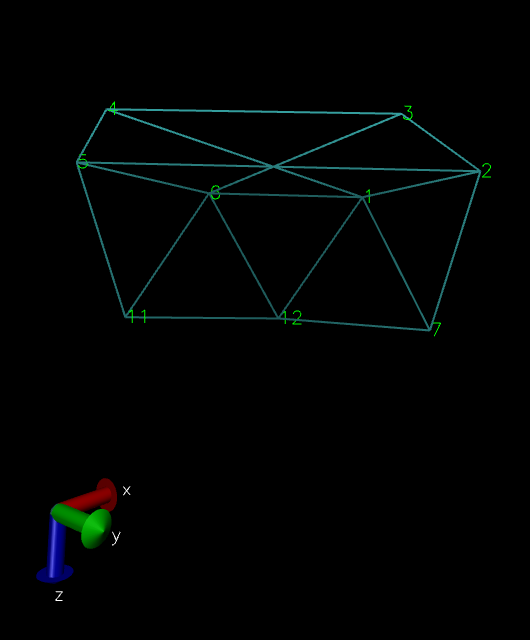
I am trying to model a CNT with triangulare lattice. Using mathematica I created the CNT’s atom positions, angles and bond information. Here is picture of it from mathematica:
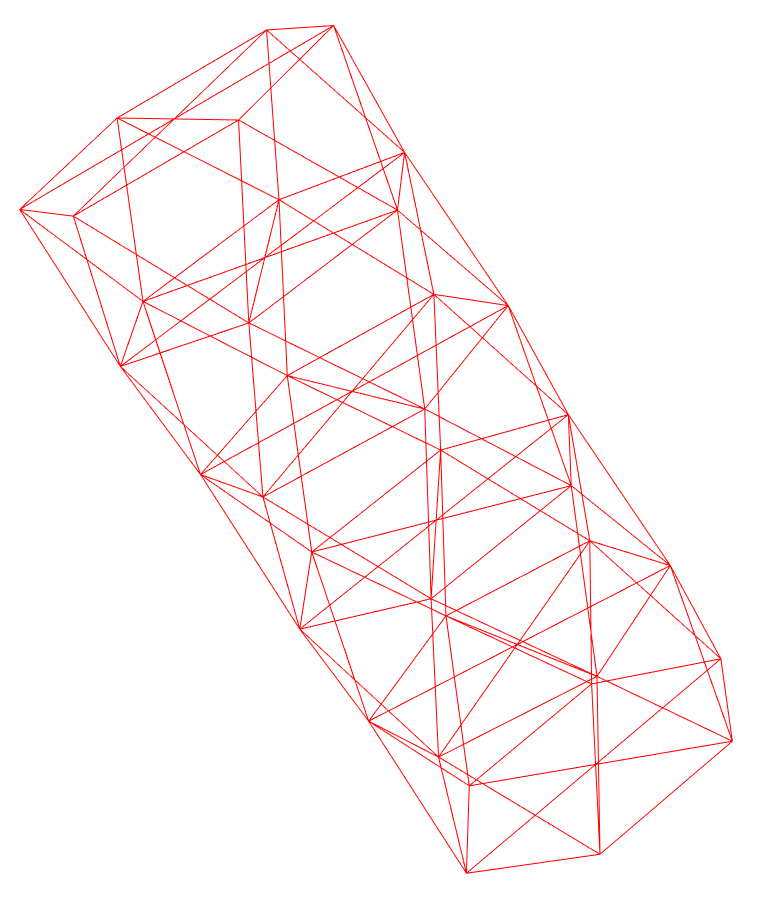
Here is the data file:
LAMMPS data file via Mathematica
36 atoms
114 bonds
180 angles
0 dihedrals
0 impropers
1 atom types
1 bond types
1 angle types
0 dihedral types
0 improper types
Masses
1 12
PairIJ Coeffs # edpd
1 1 10.75 4.5 0.41 1.58 1.42 2. 1.58
Bond Coeffs # harmonic
1 469 1.4
Angle Coeffs # harmonic
1 400. 90.
Atoms # hybrid
1 1 0.38 0 -4.3 1. 10. 0 0
2 1 0.19 0.32 -4.3 1. 10. 0 0
3 1 -0.19 0.32 -4.3 1. 10. 0 0
4 1 -0.38 0 -4.3 1. 10. 0 0
5 1 -0.19 -0.32 -4.3 1. 10. 0 0
6 1 0.19 -0.32 -4.3 1. 10. 0 0
7 1 0.32 0.19 -4. 1. 10. 0 0
8 1 0 0.38 -4. 1. 10. 0 0
9 1 -0.32 0.19 -4. 1. 10. 0 0
10 1 -0.32 -0.19 -4. 1. 10. 0 0
11 1 0 -0.38 -4. 1. 10. 0 0
12 1 0.32 -0.19 -4. 1. 10. 0 0
13 1 0.38 0 -3.6 1. 10. 0 0
14 1 0.19 0.32 -3.6 1. 10. 0 0
15 1 -0.19 0.32 -3.6 1. 10. 0 0
16 1 -0.38 0 -3.6 1. 10. 0 0
17 1 -0.19 -0.32 -3.6 1. 10. 0 0
18 1 0.19 -0.32 -3.6 1. 10. 0 0
19 1 0.32 0.19 -3.3 1. 10. 0 0
20 1 0 0.38 -3.3 1. 10. 0 0
21 1 -0.32 0.19 -3.3 1. 10. 0 0
22 1 -0.32 -0.19 -3.3 1. 10. 0 0
23 1 0 -0.38 -3.3 1. 10. 0 0
24 1 0.32 -0.19 -3.3 1. 10. 0 0
25 1 0.38 0 -3. 1. 10. 0 0
26 1 0.19 0.32 -3. 1. 10. 0 0
27 1 -0.19 0.32 -3. 1. 10. 0 0
28 1 -0.38 0 -3. 1. 10. 0 0
29 1 -0.19 -0.32 -3. 1. 10. 0 0
30 1 0.19 -0.32 -3. 1. 10. 0 0
31 1 0.32 0.19 -2.6 1. 10. 0 0
32 1 0 0.38 -2.6 1. 10. 0 0
33 1 -0.32 0.19 -2.6 1. 10. 0 0
34 1 -0.32 -0.19 -2.6 1. 10. 0 0
35 1 0 -0.38 -2.6 1. 10. 0 0
36 1 0.32 -0.19 -2.6 1. 10. 0 0
Bonds # harmonic
1 1 1 2
2 1 1 6
3 1 1 7
4 1 1 12
5 1 2 3
6 1 2 7
7 1 2 8
8 1 3 4
9 1 3 8
10 1 3 9
11 1 4 5
12 1 4 9
13 1 4 10
14 1 5 6
15 1 5 10
16 1 5 11
17 1 6 11
18 1 6 12
19 1 7 8
20 1 7 12
21 1 7 13
22 1 7 14
23 1 8 9
24 1 8 14
25 1 8 15
26 1 9 10
27 1 9 15
28 1 9 16
29 1 10 11
30 1 10 16
31 1 10 17
32 1 11 12
33 1 11 17
34 1 11 18
35 1 12 13
36 1 12 18
37 1 13 14
38 1 13 18
39 1 13 19
40 1 13 24
41 1 14 15
42 1 14 19
43 1 14 20
44 1 15 16
45 1 15 20
46 1 15 21
47 1 16 17
48 1 16 21
49 1 16 22
50 1 17 18
51 1 17 22
52 1 17 23
53 1 18 23
54 1 18 24
55 1 19 20
56 1 19 24
57 1 19 25
58 1 19 26
59 1 20 21
60 1 20 26
61 1 20 27
62 1 21 22
63 1 21 27
64 1 21 28
65 1 22 23
66 1 22 28
67 1 22 29
68 1 23 24
69 1 23 29
70 1 23 30
71 1 24 25
72 1 24 30
73 1 25 26
74 1 25 30
75 1 25 31
76 1 25 36
77 1 26 27
78 1 26 31
79 1 26 32
80 1 27 28
81 1 27 32
82 1 27 33
83 1 28 29
84 1 28 33
85 1 28 34
86 1 29 30
87 1 29 34
88 1 29 35
89 1 30 35
90 1 30 36
91 1 31 32
92 1 31 36
93 1 32 33
94 1 33 34
95 1 34 35
96 1 35 36
97 1 1 4
98 1 2 5
99 1 3 6
100 1 7 10
101 1 8 11
102 1 9 12
103 1 13 16
104 1 14 17
105 1 15 18
106 1 19 22
107 1 20 23
108 1 21 24
109 1 25 28
110 1 26 29
111 1 27 30
112 1 31 34
113 1 32 35
114 1 33 36
Angles
1 1 2 1 7
2 1 6 1 12
3 1 7 1 12
4 1 1 2 7
5 1 3 2 8
6 1 7 2 8
7 1 2 3 8
8 1 4 3 9
9 1 8 3 9
10 1 3 4 9
11 1 5 4 10
12 1 9 4 10
13 1 4 5 10
14 1 6 5 11
15 1 10 5 11
16 1 1 6 12
17 1 5 6 11
18 1 11 6 12
19 1 1 7 2
20 1 1 7 12
21 1 2 7 8
22 1 8 7 14
23 1 12 7 13
24 1 13 7 14
25 1 2 8 3
26 1 2 8 7
27 1 3 8 9
28 1 7 8 14
29 1 9 8 15
30 1 14 8 15
31 1 3 9 4
32 1 3 9 8
33 1 4 9 10
34 1 8 9 15
35 1 10 9 16
36 1 15 9 16
37 1 4 10 5
38 1 4 10 9
39 1 5 10 11
40 1 9 10 16
41 1 11 10 17
42 1 16 10 17
43 1 5 11 6
44 1 5 11 10
45 1 6 11 12
46 1 10 11 17
47 1 12 11 18
48 1 17 11 18
49 1 1 12 6
50 1 1 12 7
51 1 6 12 11
52 1 7 12 13
53 1 11 12 18
54 1 13 12 18
55 1 7 13 12
56 1 7 13 14
57 1 12 13 18
58 1 14 13 19
59 1 18 13 24
60 1 19 13 24
61 1 7 14 8
62 1 7 14 13
63 1 8 14 15
64 1 13 14 19
65 1 15 14 20
66 1 19 14 20
67 1 8 15 9
68 1 8 15 14
69 1 9 15 16
70 1 14 15 20
71 1 16 15 21
72 1 20 15 21
73 1 9 16 10
74 1 9 16 15
75 1 10 16 17
76 1 15 16 21
77 1 17 16 22
78 1 21 16 22
79 1 10 17 11
80 1 10 17 16
81 1 11 17 18
82 1 16 17 22
83 1 18 17 23
84 1 22 17 23
85 1 11 18 12
86 1 11 18 17
87 1 12 18 13
88 1 13 18 24
89 1 17 18 23
90 1 23 18 24
91 1 13 19 14
92 1 13 19 24
93 1 14 19 20
94 1 20 19 26
95 1 24 19 25
96 1 25 19 26
97 1 14 20 15
98 1 14 20 19
99 1 15 20 21
100 1 19 20 26
101 1 21 20 27
102 1 26 20 27
103 1 15 21 16
104 1 15 21 20
105 1 16 21 22
106 1 20 21 27
107 1 22 21 28
108 1 27 21 28
109 1 16 22 17
110 1 16 22 21
111 1 17 22 23
112 1 21 22 28
113 1 23 22 29
114 1 28 22 29
115 1 17 23 18
116 1 17 23 22
117 1 18 23 24
118 1 22 23 29
119 1 24 23 30
120 1 29 23 30
121 1 13 24 18
122 1 13 24 19
123 1 18 24 23
124 1 19 24 25
125 1 23 24 30
126 1 25 24 30
127 1 19 25 24
128 1 19 25 26
129 1 24 25 30
130 1 26 25 31
131 1 30 25 36
132 1 31 25 36
133 1 19 26 20
134 1 19 26 25
135 1 20 26 27
136 1 25 26 31
137 1 27 26 32
138 1 31 26 32
139 1 20 27 21
140 1 20 27 26
141 1 21 27 28
142 1 26 27 32
143 1 28 27 33
144 1 32 27 33
145 1 21 28 22
146 1 21 28 27
147 1 22 28 29
148 1 27 28 33
149 1 29 28 34
150 1 33 28 34
151 1 22 29 23
152 1 22 29 28
153 1 23 29 30
154 1 28 29 34
155 1 30 29 35
156 1 34 29 35
157 1 23 30 24
158 1 23 30 29
159 1 24 30 25
160 1 25 30 36
161 1 29 30 35
162 1 35 30 36
163 1 25 31 26
164 1 25 31 36
165 1 26 31 32
166 1 26 32 27
167 1 26 32 31
168 1 27 32 33
169 1 27 33 28
170 1 27 33 32
171 1 28 33 34
172 1 28 34 29
173 1 28 34 33
174 1 29 34 35
175 1 29 35 30
176 1 29 35 34
177 1 30 35 36
178 1 25 36 30
179 1 25 36 31
180 1 30 36 35
And here is my input file:
units lj
dimension 3
boundary p p p
neighbor 0.2 bin
neigh_modify every 1 delay 0 check yes
neigh_modify one 1500
atom_style hybrid edpd full
bond_style harmonic
angle_style harmonic
pair_style edpd 1.58 9872598
read_data cntallshort.dat
region edpd block -20 20 -20 20 -20 20 units box
#grouping
group CNT type 1
mass 1 12
set atom * edpd/temp 1.0
set atom * edpd/cv 1.0E5
pair_coeff 1 1 15.0 4.5 0.41 1.58 1.41E-5 2.0 1.58 #carbon-carbon (C-C)
compute mythermo all temp
thermo 100
thermo_modify temp mythermo
thermo_modify flush yes
comm_modify vel yes cutoff 5
#fix rigid CNT rigid molecule
velocity all create 1.0 432982 loop local dist gaussian
timestep 0.01
run 500
reset_timestep 0
compute temp all edpd/temp/atom
compute ccT all chunk/atom bin/1d y 0.0 1.0
fix stat all ave/chunk 1 500 500 ccT c_temp density/number norm sample file temp.profile
dump mydmp all atom 10 cnt.lammpstrj
#minimize 1.0e-4 1.0e-6 100 1000
run 500
The CNT I get after running my code looks like:
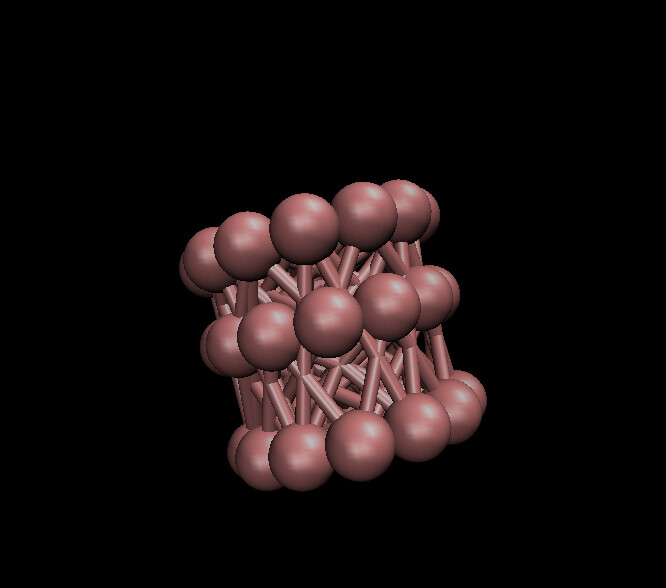


and I am getting these warning messages which even though I read through the previous question people had , I have a hard time understanding how to fix them. Could you please help me with this?
WARNING: Inconsistent image flags (src/domain.cpp:819)
WARNING: Bond/angle/dihedral extent > half of periodic box length (src/domain.cpp:940)
Per MPI rank memory allocation (min/avg/max) = 34.64 | 34.64 | 34.64 Mbytes
Step Temp E_pair E_mol TotEng Press
0 1 268.81913 2076.5914 2346.8689 26063.561
100 1 268.81913 2076.5914 2346.8689 25212.867
200 1 268.81913 2076.5914 2346.8689 26729.651
300 1 268.81913 2076.5914 2346.8689 26359.631
400 1 268.81913 2076.5914 2346.8689 24243.61
500 1 268.81913 2076.5914 2346.8689 26871.991