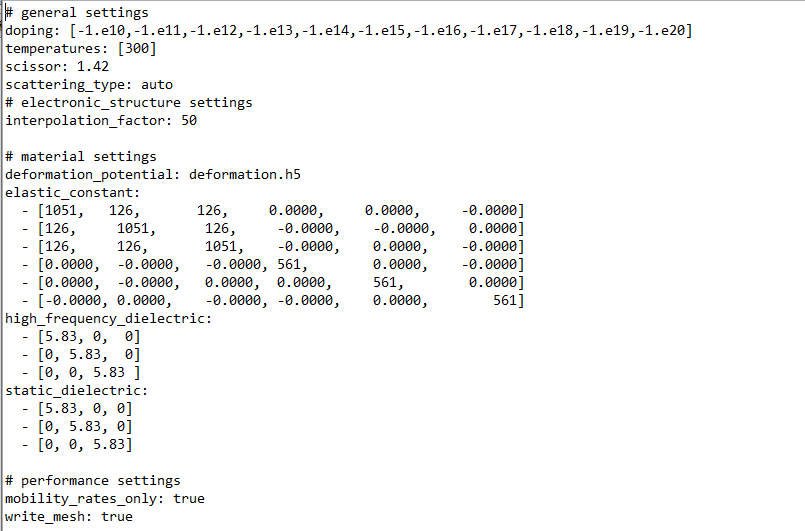
When I used diamond as the calculation material to calculate the mobility, there was a big error, but the cause has not been found yet, could you please give me some guidance @alex
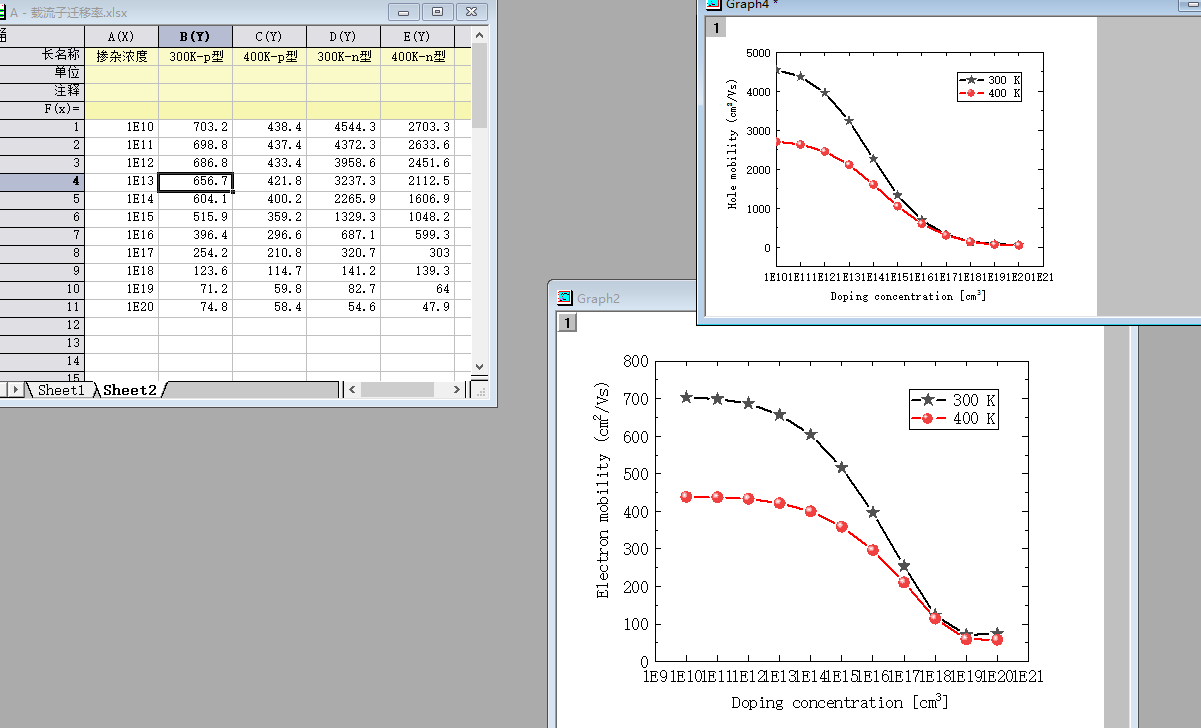
This is my calculation of diamond mobility, but according to the literature, the electronic mobility of diamond at room temperature is about 4000cm2V-1S-1, which obviously does not meet the requirements
Thank you very much for your reply
On the first question; The result has converged. This picture is the electron and hole mobility of diamond calculated by me. The error in the calculation of hole mobility is small on the surface of the calculation result, but it is large in the calculation of electron mobility
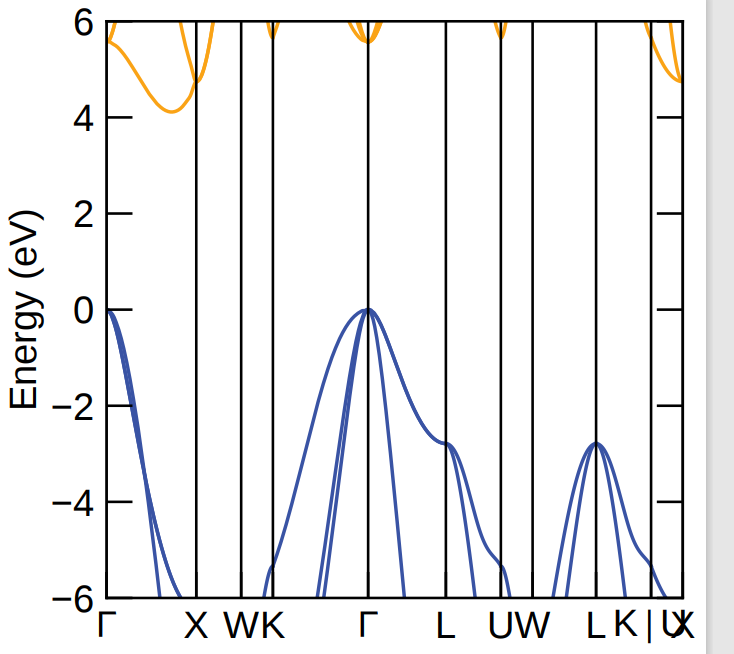
I’ve been working on this lately, and I saw a command in the forum: amset plot band vasprun.xml. So I used this to extract the image, as shown below; Does this mean that when you calculate the mobility of electrons you don’t have enough bands, and this is my guess,