Dear Alex and kind users,
When I use crystal cell and crystal cell to calculate the mobility of diamond system, their results are very different. I get ‘wavefunction.h5’ and 'deformation.H5 ’ with crystal cell and unit cell respectively. When the doping concentration is 1e16, the result of crystal cell is 1552 cm ^ 2 / Vs and that of crystal cell is 454 cm ^ 2 /Vs,the result of the experiment is about 2000 cm ^ 2 /Vs. Is it normal to have such a big difference?
Hi @chenjiashu, can I double check:
- What k-point mesh did you use for the wavefunction.h5 and deformation.h5 for the primitive and unit cells.
- What k-point mesh did you use for the vasprun.xml for the primitive and unit cells.
- Did you converge the transport results with respect to interpolation_factor in both cases? E.g., keep increasing interpolation_factor until the mobility stops changing by much?
Best,
Alex
1 Like
Dear Alex and kind users,
- I used 12X12X12( primitive cells) and 7X7X7(unit cells) k-point mesh for the wavefunction.h5, deformation.h5. For primitive cells (a=b=c=2.527Å,α=β=γ=60°), unit cells (a=b=c=3.567Å,α=β=γ=90°)
- I used 12X12X12( primitive cells) and 7X7X7(unit cells) k-point mesh for the vasprun.xml.
primitive cell:
interpolation_factor =60, mobility = 350.1 (doping: -1.00e+16)
interpolation_factor =80, mobility = 353.7 (doping: -1.00e+16)
interpolation_factor =100, mobility = 357.4 (doping: -1.00e+16)
conventional unitcell:
interpolation_factor =60, mobility = 1544.4 (doping: -1.00e+16)
interpolation_factor =80, mobility = 1552.3 (doping: -1.00e+16)
interpolation_factor =100, mobility = 1609.4 (doping: -1.00e+16)
Thanks for the information. This is very unusual.
Please can you send the amset.log file for each structure and also the output of amset deform read for each structure.
Dear Alex and kind users,
The output of amset deform read for each structure are:
- deformation(primitive cell).h5 (327.2 KB)
-
deformation(unit cell).h5 (96.2 KB)
respectively.
amset.log file for primitive cell is
/$$$$$$ /$$ /$$ /$$$$$$ /$$$$$$$$ /$$$$$$$$
/$$__ $$| $$$ /$$$ /$$__ $$| $$_____/|__ $$__/
| $$ \ $$| $$$$ /$$$$| $$ \__/| $$ | $$
| $$$$$$$$| $$ $$/$$ $$| $$$$$$ | $$$$$ | $$
| $$__ $$| $$ $$$| $$ \____ $$| $$__/ | $$
| $$ | $$| $$\ $ | $$ /$$ \ $$| $$ | $$
| $$ | $$| $$ \/ | $$| $$$$$$/| $$$$$$$$ | $$
|__/ |__/|__/ |__/ \______/ |________/ |__/
v0.4.15
Ganose, A. M., Park, J., Faghaninia, A., Woods-Robinson,
R., Persson, K. A., Jain, A. Efficient calculation of
carrier scattering rates from first principles.
Nat. Commun. 12, 2222 (2021)
amset starting on 11 Apr 2022 at 17:57
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ SETTINGS ~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Run parameters:
- scattering_type: ['IMP', 'ADP']
- doping: [-1.e+14 -1.e+15 -1.e+16 -1.e+17 -1.e+18]
- temperatures: [300]
- bandgap: 5.48
- soc: False
- zero_weighted_kpoints: prefer
- interpolation_factor: 80
- wavefunction_coefficients: wavefunction.h5
- use_projections: False
- free_carrier_screening: False
- high_frequency_dielectric:
[[ 5.82 0.00 0.00]
[ 0.00 5.82 0.00]
[ 0.00 0.00 5.82]]
- static_dielectric:
[[ 5.82 0.00 0.00]
[ 0.00 5.82 0.00]
[ 0.00 0.00 5.82]]
- elastic_constant:
[[1060.8 130.7 130.7 0.0 0.0 0.0]
[ 130.7 1060.8 130.7 0.0 0.0 0.0]
[ 130.7 130.7 1060.8 0.0 0.0 0.0]
[ 0.0 0.0 0.0 567.0 0.0 0.0]
[ 0.0 0.0 0.0 0.0 567.0 0.0]
[ 0.0 0.0 0.0 0.0 0.0 567.0]]
- deformation_potential: deformation.h5
- defect_charge: 1
- compensation_factor: 2
- pop_frequency: 38.95
- energy_cutoff: 1.5
- fd_tol: 0.05
- dos_estep: 0.01
- symprec: 0.01
- nworkers: 24
- cache_wavefunction: True
- calculate_mobility: True
- separate_mobility: True
- mobility_rates_only: False
- file_format: json
- write_input: False
- write_mesh: True
- print_log: True
- write_log: True
~~~~~~~~~~~~~~~~~~~~~~~~~~~~ STRUCTURE ~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Structure information:
- formula: C
- # sites: 2
- space group: Fd3-m
Lattice:
- a, b, c [angstrom]: 2.52, 2.52, 2.52
- a, b, y [deg]: 60, 60, 60
~~~~~~~~~~~~~~~~~~~~~~~~~~ BAND STRUCTURE ~~~~~~~~~~~~~~~~~~~~~~~~~
Input band structure information:
- # bands: 12
- # k-points: 868
- Fermi level: 9.756 eV
- spin polarized: False
- metallic: False
Band gap:
- indirect band gap: 4.152 eV
- direct band gap: 5.590 eV
- direct k-point: [0.00, 0.00, 0.00]
Valence band maximum:
- energy: 9.727 eV
- k-point: [0.00, 0.00, 0.00]
- band indices: 2, 3, 4
Conduction band minimum:
- energy: 13.880 eV
- k-point: [0.33, 0.33, 0.00]
- band indices: 5
~~~~~~~~~~~~~~~~~~~~~~~~~~ INTERPOLATION ~~~~~~~~~~~~~~~~~~~~~~~~~~
Getting band interpolation coefficients
- time: 285.6886 s
Interpolation parameters:
- k-point mesh: 203x203x203
- energy cutoff: 1.5 eV
Interpolating spin-up bands 2-7
- time: 25.1521 s
bandgap set to 5.480 eV, applying scissor of 1.338 eV
Generating tetrahedron mesh vertices
- time: 77.2374 s
Initializing tetrahedron band structure
- time: 43.6057 s
Initializing momentum relaxation time factor calculator
Initializing wavefunction overlap calculator
Desymmetrizing k-point mesh
- Found initial mesh: 12.000 x 12.000 x 12.000
- Integer mesh: 12 x 12 x 12
- Using 48 symmetry operations
Desymmetrizing wavefunction coefficients
- time: 0.1598 s
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ DOS ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
DOS parameters:
- emin: -4.31 eV
- emax: 27.20 eV
- dos weight: 2
- n points: 3151
Generating tetrahedral DOS:
- time: 205.6955 s
Intrinsic DOS Fermi level: 11.7983 eV
DOS contains 6.000 electrons
Calculated Fermi levels:
conc [cm-3] temp [K] E_fermi [eV]
------------- ---------- --------------
-1.00e+14 300.0 14.1957
-1.00e+15 300.0 14.2553
-1.00e+16 300.0 14.3148
-1.00e+17 300.0 14.3743
-1.00e+18 300.0 14.4340
Calculated Fermi-Dirac cut-offs:
- min: 14.537 eV
- max: 14.747 eV
~~~~~~~~~~~~~~~~~~~~~~~~~~~~ SCATTERING ~~~~~~~~~~~~~~~~~~~~~~~~~~~
Scattering mechanisms to be calculated: IMP, ADP
Inverse screening length (b) and impurity concentration (N_i):
conc [cm-3] temp [K] b2 [a^-2] N_i [cm-3]
------------- ---------- ----------- ------------
-1.00e+14 300.0 3.37e-08 2.00e+14
-1.00e+15 300.0 3.37e-07 2.00e+15
-1.00e+16 300.0 3.37e-06 2.00e+16
-1.00e+17 300.0 3.37e-05 2.00e+17
-1.00e+18 300.0 3.35e-04 2.00e+18
Initializing deformation potential interpolator
Forking 24 processes to calculate scattering
- time: 36.3252 s
Scattering information:
- # ir k-points: 182104
Calculating rates for spin-up band 1
- # k-points within Fermi-Dirac cut-offs: 0
- time: 1.7218 s
Calculating rates for spin-up band 2
- # k-points within Fermi-Dirac cut-offs: 0
- time: 1.3365 s
Calculating rates for spin-up band 3
- # k-points within Fermi-Dirac cut-offs: 0
- time: 1.8674 s
Calculating rates for spin-up band 4
- # k-points within Fermi-Dirac cut-offs: 71226
- time: 266.6401 s
Calculating rates for spin-up band 5
- # k-points within Fermi-Dirac cut-offs: 0
- time: 1.2177 s
Calculating rates for spin-up band 6
- # k-points within Fermi-Dirac cut-offs: 0
- time: 1.1981 s
Interpolating missing scattering rates
- time: 1.0529 s
Filling scattering rates [s⁻¹] outside FD cutoffs with:
conc [cm-3] temp [K] IMP ADP
------------- ---------- -------- --------
-1.00e+14 300.0 6.47e+10 1.32e+13
-1.00e+15 300.0 2.16e+11 1.32e+13
-1.00e+16 300.0 8.02e+11 1.32e+13
-1.00e+17 300.0 3.66e+12 1.32e+13
-1.00e+18 300.0 1.70e+13 1.32e+13
~~~~~~~~~~~~~~~~~~~~~~~~~~~~ TRANSPORT ~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Calculating conductivity, Seebeck, and electronic thermal
conductivity
- time: 215.3347 s
Calculating overall mobility
- time: 197.7521 s
Calculating individual scattering rate mobilities
- time: 392.1610 s
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ RESULTS ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Average conductivity (σ), Seebeck (S) and mobility (μ) results:
conc [cm-3] temp [K] σ [S/m] S [µV/K] μ [cm2/Vs]
------------- ---------- --------- ---------- ------------
-1.00e+14 300.0 7.72e-01 -1.32e+03 481.8
-1.00e+15 300.0 6.98e+00 -1.14e+03 435.5
-1.00e+16 300.0 5.67e+01 -9.55e+02 353.7
-1.00e+17 300.0 3.60e+02 -7.86e+02 224.8
-1.00e+18 300.0 1.60e+03 -6.15e+02 100.1
Mobility breakdown by scattering mechanism, in cm2/Vs:
conc [cm-3] temp [K] IMP ADP
------------- ---------- -------- --------
-1.00e+14 300.0 4.74e+04 5.22e+02
-1.00e+15 300.0 1.41e+04 5.22e+02
-1.00e+16 300.0 3.70e+03 5.22e+02
-1.00e+17 300.0 7.79e+02 5.22e+02
-1.00e+18 300.0 1.59e+02 5.21e+02
Results written to:
/home/smc/mobility/C/transport_203x203x203.json
and
/home/smc/mobility/C/mesh_203x203x203.h5
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ END ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Timing and memory usage:
- interpolation time: 457.0914 s
- dos time: 249.0441 s
- scattering time: 335.8715 s
- transport time: 805.2606 s
- writing time: 17.5555 s
- total time: 1877.8705 s
- max memory: 76888.4 MB
amset exiting on 11 Apr 2022 at 18:28
amset.log file for unit cell is
/$$$$$$ /$$ /$$ /$$$$$$ /$$$$$$$$ /$$$$$$$$
/$$__ $$| $$$ /$$$ /$$__ $$| $$_____/|__ $$__/
| $$ \ $$| $$$$ /$$$$| $$ \__/| $$ | $$
| $$$$$$$$| $$ $$/$$ $$| $$$$$$ | $$$$$ | $$
| $$__ $$| $$ $$$| $$ \____ $$| $$__/ | $$
| $$ | $$| $$\ $ | $$ /$$ \ $$| $$ | $$
| $$ | $$| $$ \/ | $$| $$$$$$/| $$$$$$$$ | $$
|__/ |__/|__/ |__/ \______/ |________/ |__/
v0.4.15
Ganose, A. M., Park, J., Faghaninia, A., Woods-Robinson,
R., Persson, K. A., Jain, A. Efficient calculation of
carrier scattering rates from first principles.
Nat. Commun. 12, 2222 (2021)
amset starting on 09 Apr 2022 at 18:19
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ SETTINGS ~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Run parameters:
- scattering_type: auto
- doping: [-1.e+14 -1.e+15 -1.e+16 -1.e+17 -1.e+18]
- temperatures: [300]
- bandgap: 5.5
- soc: False
- zero_weighted_kpoints: prefer
- interpolation_factor: 80
- wavefunction_coefficients: wavefunction.h5
- use_projections: False
- free_carrier_screening: False
- high_frequency_dielectric:
[[ 5.82 -0.00 0.00]
[ 0.00 5.82 0.00]
[ -0.00 0.00 5.82]]
- static_dielectric:
[[ 5.82 -0.00 0.00]
[ 0.00 5.82 0.00]
[ -0.00 0.00 5.82]]
- elastic_constant:
[[1060.8 130.7 130.7 0.0 0.0 0.0]
[ 130.7 1060.8 130.7 0.0 0.0 0.0]
[ 130.7 130.7 1060.8 0.0 0.0 0.0]
[ 0.0 0.0 0.0 567.0 0.0 0.0]
[ 0.0 0.0 0.0 0.0 567.0 0.0]
[ 0.0 0.0 0.0 0.0 0.0 567.0]]
- deformation_potential: deformation.h5
- defect_charge: 1
- compensation_factor: 2
- energy_cutoff: 1.5
- fd_tol: 0.05
- dos_estep: 0.01
- symprec: 0.01
- nworkers: -1
- cache_wavefunction: True
- calculate_mobility: True
- separate_mobility: True
- mobility_rates_only: False
- file_format: json
- write_input: False
- write_mesh: True
- print_log: True
- write_log: True
~~~~~~~~~~~~~~~~~~~~~~~~~~~~ STRUCTURE ~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Structure information:
- formula: C
- # sites: 8
- space group: Fd3-m
Lattice:
- a, b, c [angstrom]: 3.57, 3.57, 3.57
- a, b, y [deg]: 90, 90, 90
~~~~~~~~~~~~~~~~~~~~~~~~~~ BAND STRUCTURE ~~~~~~~~~~~~~~~~~~~~~~~~~
Input band structure information:
- # bands: 24
- # k-points: 172
- Fermi level: 9.770 eV
- spin polarized: False
- metallic: False
Band gap:
- indirect band gap: 4.129 eV
- direct band gap: 4.717 eV
- direct k-point: [0.14, 0.00, 0.00]
Valence band maximum:
- energy: 9.727 eV
- k-point: [0.00, 0.00, 0.00]
- band indices: 14, 15, 16
Conduction band minimum:
- energy: 13.856 eV
- k-point: [0.29, 0.00, 0.00]
- band indices: 17
~~~~~~~~~~~~~~~~~~~~~~~~~~ INTERPOLATION ~~~~~~~~~~~~~~~~~~~~~~~~~~
Getting band interpolation coefficients
- time: 13.8244 s
Interpolation parameters:
- k-point mesh: 109x109x109
- energy cutoff: 1.5 eV
Interpolating spin-up bands 14-24
- time: 7.9275 s
bandgap set to 5.500 eV, applying scissor of 1.373 eV
Generating tetrahedron mesh vertices
- time: 11.8100 s
Initializing tetrahedron band structure
- time: 27.0868 s
Initializing momentum relaxation time factor calculator
Initializing wavefunction overlap calculator
Desymmetrizing k-point mesh
- Found initial mesh: 7.000 x 7.000 x 7.000
- Integer mesh: 7 x 7 x 7
- Using 48 symmetry operations
Desymmetrizing wavefunction coefficients
- time: 0.1382 s
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ DOS ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
DOS parameters:
- emin: 3.79 eV
- emax: 22.39 eV
- dos weight: 2
- n points: 1860
Generating tetrahedral DOS:
- time: 160.4284 s
Intrinsic DOS Fermi level: 11.7906 eV
DOS contains 6.000 electrons
Calculated Fermi levels:
conc [cm-3] temp [K] E_fermi [eV]
------------- ---------- --------------
-1.00e+14 300.0 14.2146
-1.00e+15 300.0 14.2741
-1.00e+16 300.0 14.3337
-1.00e+17 300.0 14.3932
-1.00e+18 300.0 14.4530
Calculated Fermi-Dirac cut-offs:
- min: 14.544 eV
- max: 14.764 eV
~~~~~~~~~~~~~~~~~~~~~~~~~~~~ SCATTERING ~~~~~~~~~~~~~~~~~~~~~~~~~~~
Examining material properties to determine possible scattering
mechanisms
Scattering mechanisms to be calculated: ADP, IMP
Initializing deformation potential interpolator
Inverse screening length (b) and impurity concentration (N_i):
conc [cm-3] temp [K] b2 [a^-2] N_i [cm-3]
------------- ---------- ----------- ------------
-1.00e+14 300.0 3.37e-08 2.00e+14
-1.00e+15 300.0 3.37e-07 2.00e+15
-1.00e+16 300.0 3.37e-06 2.00e+16
-1.00e+17 300.0 3.36e-05 2.00e+17
-1.00e+18 300.0 3.33e-04 2.00e+18
Forking 24 processes to calculate scattering
- time: 11.9195 s
Scattering information:
- # ir k-points: 29260
Calculating rates for spin-up band 1
- # k-points within Fermi-Dirac cut-offs: 0
- time: 1.0252 s
Calculating rates for spin-up band 2
- # k-points within Fermi-Dirac cut-offs: 0
- time: 0.6214 s
Calculating rates for spin-up band 3
- # k-points within Fermi-Dirac cut-offs: 0
- time: 0.3188 s
Calculating rates for spin-up band 4
- # k-points within Fermi-Dirac cut-offs: 37290
- time: 338.1230 s
Calculating rates for spin-up band 5
- # k-points within Fermi-Dirac cut-offs: 0
- time: 0.2774 s
Calculating rates for spin-up band 6
- # k-points within Fermi-Dirac cut-offs: 0
- time: 0.2694 s
Calculating rates for spin-up band 7
- # k-points within Fermi-Dirac cut-offs: 0
- time: 0.2714 s
Calculating rates for spin-up band 8
- # k-points within Fermi-Dirac cut-offs: 0
- time: 0.2594 s
Calculating rates for spin-up band 9
- # k-points within Fermi-Dirac cut-offs: 0
- time: 0.2631 s
Calculating rates for spin-up band 10
- # k-points within Fermi-Dirac cut-offs: 0
- time: 0.2639 s
Calculating rates for spin-up band 11
- # k-points within Fermi-Dirac cut-offs: 0
- time: 0.2584 s
Interpolating missing scattering rates
- time: 0.4565 s
Filling scattering rates [s⁻¹] outside FD cutoffs with:
conc [cm-3] temp [K] ADP IMP
------------- ---------- -------- --------
-1.00e+14 300.0 2.40e+12 9.37e+10
-1.00e+15 300.0 2.40e+12 3.09e+11
-1.00e+16 300.0 2.40e+12 1.10e+12
-1.00e+17 300.0 2.40e+12 4.62e+12
-1.00e+18 300.0 2.40e+12 1.94e+13
~~~~~~~~~~~~~~~~~~~~~~~~~~~~ TRANSPORT ~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Calculating conductivity, Seebeck, and electronic thermal
conductivity
- time: 71.0429 s
Calculating overall mobility
- time: 70.4653 s
Calculating individual scattering rate mobilities
- time: 139.0019 s
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ RESULTS ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Average conductivity (σ), Seebeck (S) and mobility (μ) results:
conc [cm-3] temp [K] σ [S/m] S [µV/K] μ [cm2/Vs]
------------- ---------- --------- ---------- ------------
-1.00e+14 300.0 6.31e+00 -1.29e+03 3940.0
-1.00e+15 300.0 4.41e+01 -1.11e+03 2755.3
-1.00e+16 300.0 2.49e+02 -9.42e+02 1552.3
-1.00e+17 300.0 9.84e+02 -7.75e+02 613.9
-1.00e+18 300.0 2.92e+03 -5.85e+02 182.1
Mobility breakdown by scattering mechanism, in cm2/Vs:
conc [cm-3] temp [K] ADP IMP
------------- ---------- -------- --------
-1.00e+14 300.0 6.23e+03 4.60e+04
-1.00e+15 300.0 6.23e+03 1.38e+04
-1.00e+16 300.0 6.23e+03 3.80e+03
-1.00e+17 300.0 6.23e+03 8.75e+02
-1.00e+18 300.0 6.19e+03 2.00e+02
Results written to:
/media/chen/0004124B00014E84/s-100-5/C_mp-66_conventional_standard/
transport_109x109x109.json
and
/media/chen/0004124B00014E84/s-100-5/C_mp-66_conventional_standard/
mesh_109x109x109.h5
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ END ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Timing and memory usage:
- interpolation time: 69.5208 s
- dos time: 173.6755 s
- scattering time: 363.1195 s
- transport time: 280.5144 s
- writing time: 1.0935 s
- total time: 908.1114 s
- max memory: 16917.1 MB
amset exiting on 09 Apr 2022 at 18:34
There seems to be an issue with the valence band maximum deformation potentials in the primitive cell structure.
For the conventional (unit) cell, the deformation potentials of the three degenerate valence bands at the gamma point are:
VBM 1:
[[7.54, 0.46, 0.46]
[0.46, 7.54, 0.46]
[0.46, 0.46, 7.54]]
VBM 2:
[[7.76, 0.3 , 0.3 ]
[0.3 , 7.76, 0.3 ]
[0.3 , 0.3 , 7.76]]
VBM 3:
[[7.55, 0.42, 0.42]
[0.42, 7.55, 0.42]
[0.42, 0.42, 7.55]]
This makes sense as the deformation potentials are all nearly degenerate and the tensors are more or less diagonal.
For the primitive cell, the picture is quite different:
VBM 1:
[[ 4.05, 8.84, 12.38]
[ 8.84, 3.77, 10.71]
[12.38, 10.71, 2.6 ]]
VBM 2:
[[0.4 , 0.1 , 0.14],
[0.1 , 5.18, 6.94],
[0.14, 6.94, 2.85]]
VBM 3:
[[ 3.88, 8.62, 12.26]
[ 8.62, 3.86, 10.8 ]
[12.26, 10.8 , 2.75]]
Clearly, something is wrong with these deformation potentials!
I’m not entirely sure how this could happen.
One potential issue is that you’re not using the standardised primitive cell representation. Specifically, the lattice parameters you’re using are:
a = 2.52225 , 0. , 0.
b = 1.261125, 2.184332, 0.
c = 1.261125, 0.728111, 2.059408
But the standardised representation (which will align the structure so that the cartesian directions align with the unit cell) is:
a = 0. , 1.7835, 1.7835
b = 1.7835, 0. , 1.7835
c = 1.7835, 1.7835, 0.
You can easily convert your structure to the standardised format using pymatgen. For example:
from pymatgen.symmetry.analyzer import SpacegroupAnalyzer
from pymatgen.core import Structure
structure = Structure.from_file("POSCAR")
sga = SpacegroupAnalyzer(structure, symprec=0.01)
primitive = sga.get_primitive_standard_structure()
primitive.to(fmt="poscar", filename="POSCAR_prim")
I’ve attached the standardised primitive structure. Please could you try recalculating the deformation potentials and send over the new deformation.h5 file.
Best,
Alex
POSCAR_prim (158 Bytes)
1 Like
Dear Alex
I use the standardised primitive structure which you give for calculation. I get ‘wavefunction.h5’ , 'deformation.H5 ’ and ‘vasprun.xml’ with unit cell and standardised primitive structure respectively. When the doping concentration is -1e16, the result of uint cell is 1552 cm ^ 2 / Vs and that of standardised primitive structure is 994.7 cm ^ 2 / Vs. There is still a big difference between the calculation of unit cell andstandardised primitive structure.
amset.log file for standardised primitive structure with different ‘interpolation_factor’ are:
amset.log-60-new (8.2 KB)
amset.log-80-new (8.1 KB)
amset.log-100-new (8.1 KB)
Please can you send me the deformation.h5 of the standardised primitive structure?
Dear Alex
The deformation.h5 of the standardised primitive structure is
deformation.h5 (304.3 KB)
Ok, using the correct primitive has fixed the deformation potentials. The deformations of the degenerate VBMs at gamma are now:
VBM 1:
[[7.52, 0.44, 0.44]
[0.44, 7.52, 0.44]
[0.44, 0.44, 7.52]]
VBM 2:
[[7.76, 0.28, 0.28]
[0.28, 7.76, 0.28]
[0.28, 0.28, 7.76]]
VBM 3:
[[7.57, 0.44, 0.44]
[0.44, 7.57, 0.44]
[0.44, 0.44, 7.57]]
So these now match the deformation potentials from the unit cell.
Just to confirm, did you also regenerate the wavefunction.h5 and vasprun.xml file using the standardised structure too?
Best,
Alex
Dear Alex
I also have got ‘wavefunction.h5’ and ‘vasprun.xml’ with standardised primitive structure.
wavefunction.h5 is
wavefunction.h5 (416.6 KB)
vasprun.xml is
vasprun.xml (3.2 MB)
Thank you very much.
I’m a little unsure of what could be causing the remaining discrepancy. Are you use you used the correct wavefunction.h5, deformation.h5, and vasprun.xml in each case?
Please could you send me the vasprun.xml file for the conventional cell too?
I just tried plotting the interpolated band structure of the primitive cell using the vasprun.xml you sent (using amset plot band --interpolation-factor 80 vasprun.xml) and it seems the interpolation of the conduction band is very poor.
You should also try increasing the k-point mesh of the vasprun.xml, wavefunction.h5, and deformation.h5 calculations. Perhaps something like 24x24x24 will give better results.
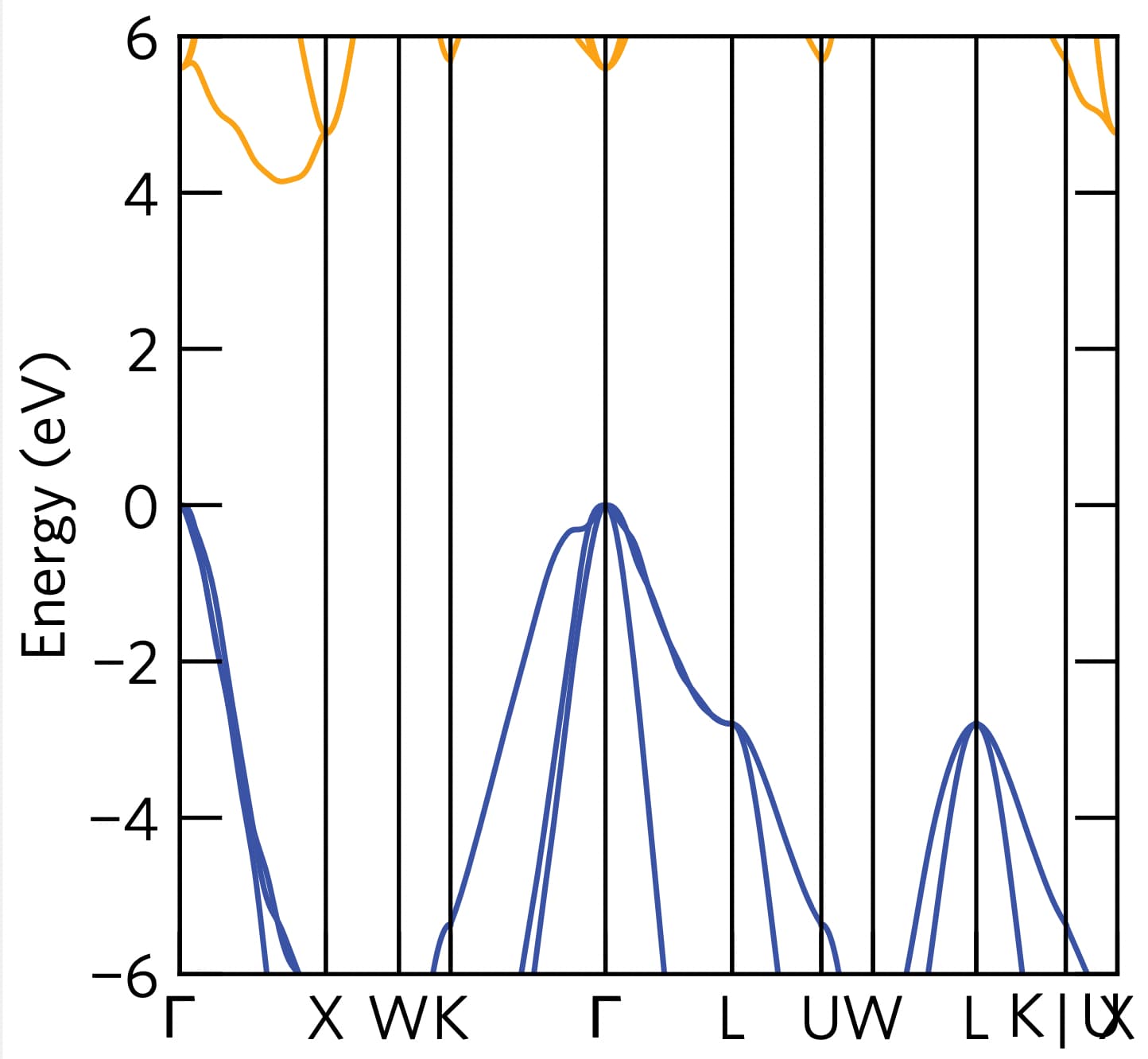
1 Like
Dear Alex
‘wavefunction.h5’ , 'deformation.H5 ’ and ‘vasprun.xml’ of conventional unit cell are
wavefunction.h5 (862.8 KB)
deformation(unit cell).h5 (96.2 KB)
vasprun.xml (3.6 MB)
And I also also try increasing the k-point mesh(24x24x24) of the vasprun.xml, wavefunction.h5, and deformation.h5 calculations. But we have not enough memory to run “python main.py” (our computer memory is 128G , we also try to set up “nworks=2” but it still doesn’t work.)
And I also also try increasing the k-point mesh(24x24x24) of the vasprun.xml, wavefunction.h5, and deformation.h5 calculations. But we have not enough memory to run “python main.py” (our computer memory is 128G , we also try to set up “nworks=2” but it still doesn’t work.)
You can try reducing the interpolation_factor down to 5 and keep increasing it by 5 until the results are roughly converged. The interpolation factor you need with a dense initial mesh will be much smaller than what you need with a coarser initial mesh.
Dear Alex
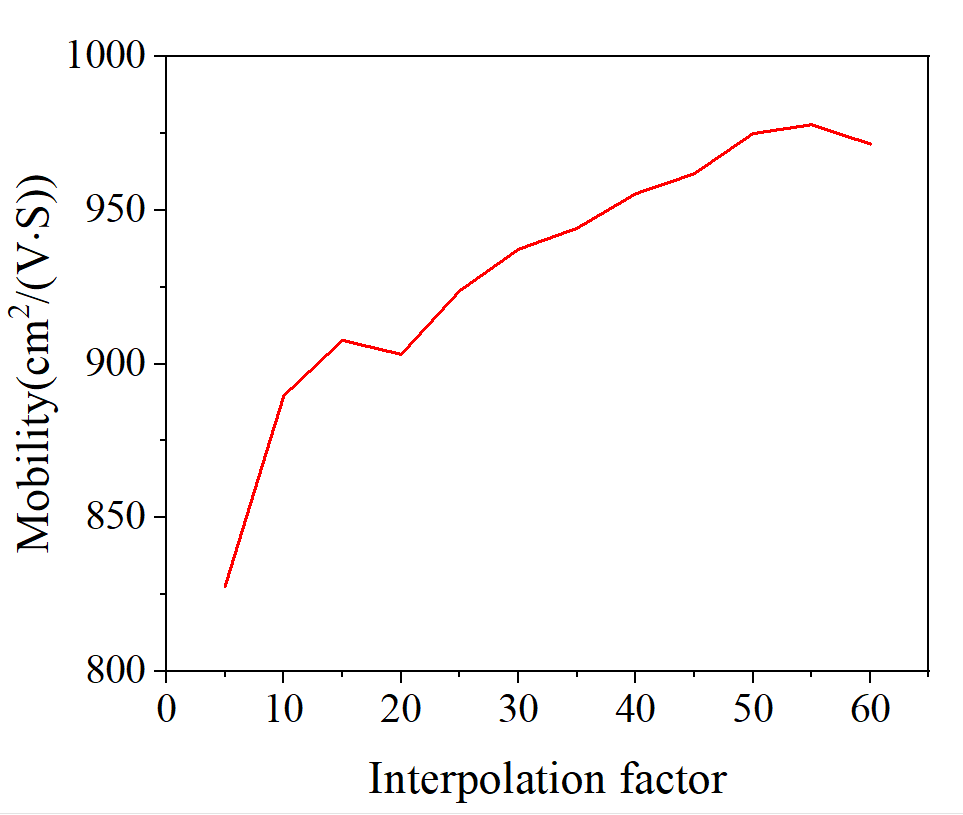
- I try reducing the interpolation_factor down to 5 and keep increasing it by 5 until interpolation_factor=60. The variation of mobility with interpolation_facto is shown as follows ( I use the standardised primitive structure which you give for calculation, and k-point mesh is 12x12x12)
2.I also also try increasing the k-point mesh(24x24x24) of the vasprun.xml, wavefunction.h5, and deformation.h5 calculations. When interpolation_facto=5( When k-point mesh is 24x24x24 and interpolation_facto>5,we have not enough memory to run “python main.py”. So we set interpolation_facto=5)the mobility of the standardised primitive structure is 833.5 cm ^ 2 / Vs ( k-point mesh is 24x24x24), 827.6 cm ^ 2 / Vs ( k-point mesh is 12x12x12). The result of uint cell is 1552 cm ^ 2 / Vs, there is still a big difference between the calculation of unit cell and the standardised primitive structure.