Since I raised the issue of an anomaly in the system’s energy after the fix evaporate in the simulation last time, I still have not figured out the issue. Therefore, I am giving a more detailed post here again, and thanks Mr. Axel for his patient guidance.
Run in LAMMPS (2 Aug 2023 - Update 1), on CentOS with 64 cores.
The simulation I am conducting is of water evaporating from a Cu surface at room temperature, as shown in the figure.
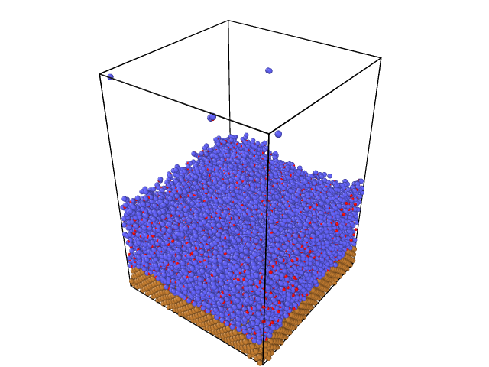
The water molecules that drift from the surface to the top are considered evaporated, and I use the fix evaporate command to remove them. Before I proceed with the evaporation, I confirm that the system has already reached thermal equilibrium.
I conducted a simulation as follows: The total number of steps in the simulation was 15 million. In the first phase, which consisted of the initial 5 million steps, I did not use fix evaporate and simply carried out a normal fix nve preheat. Then, in the second phase, I used fix evaporate for 5 million steps to simulate evaporation. In the final phase, the last 5 million steps, I turned off fix evaporate and the entire system was fixed with nve. Normally, during the fix evaporate phase, both the kinetic and potential energy of the system should gradually decrease (due to the deletion of water molecules). However, this was not the case:
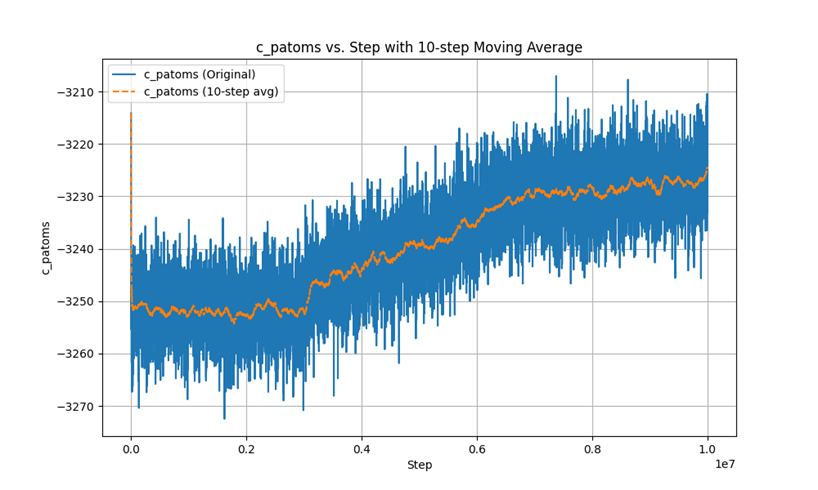
This is the Pe-steps curve.
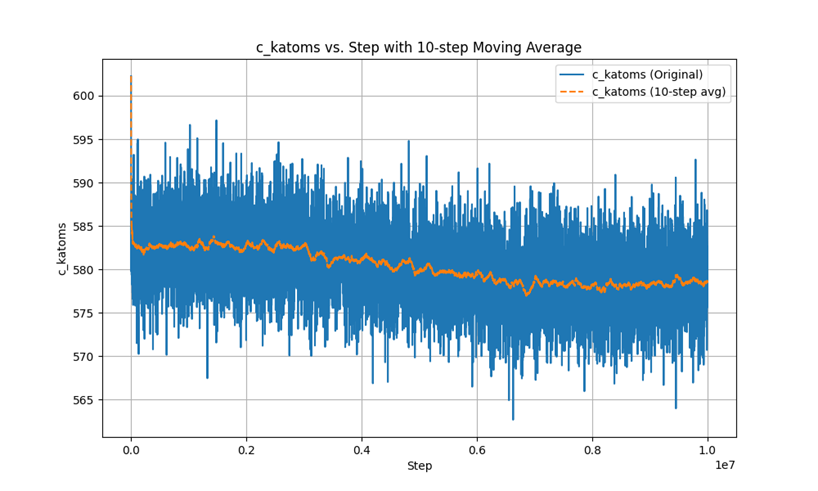
This is the Ke-steps curve.
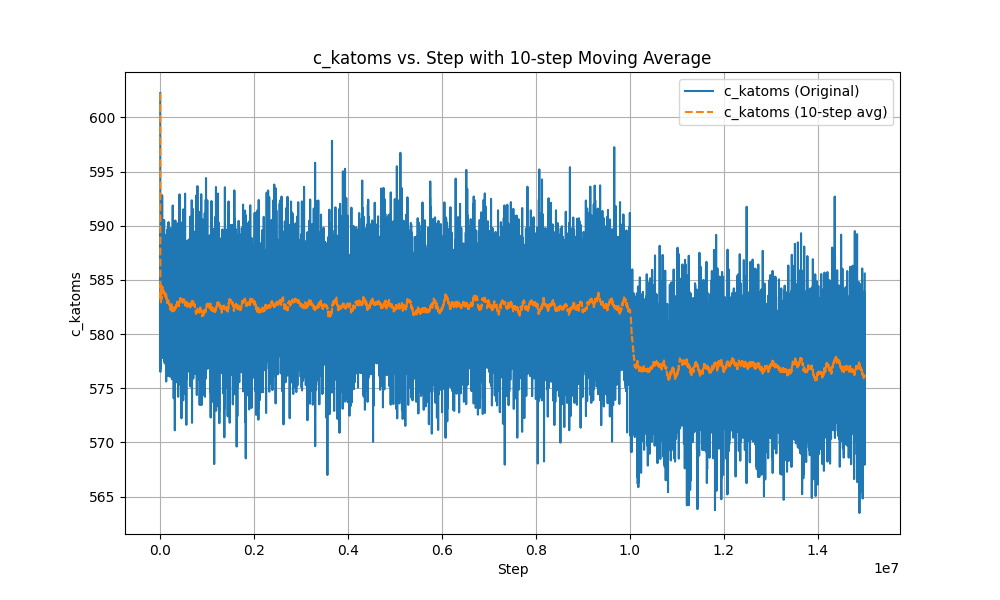
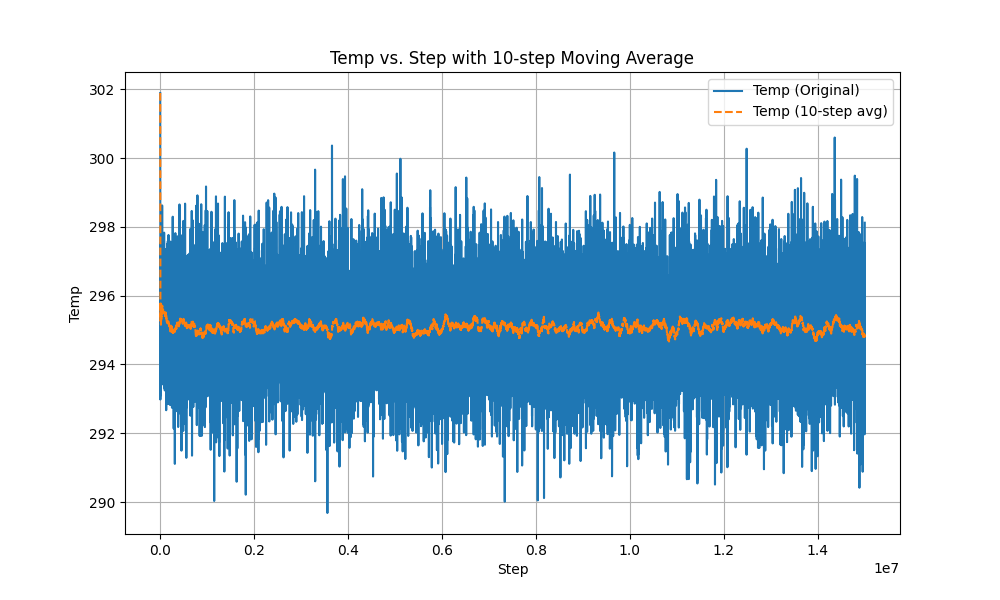
In the second phase, after using fix evaporate, the potential energy of the system decreased, but the total kinetic energy of the system did not change, which is a very contradictory result. I checked the temperature and found that the temperature did not change significantly throughout the process.
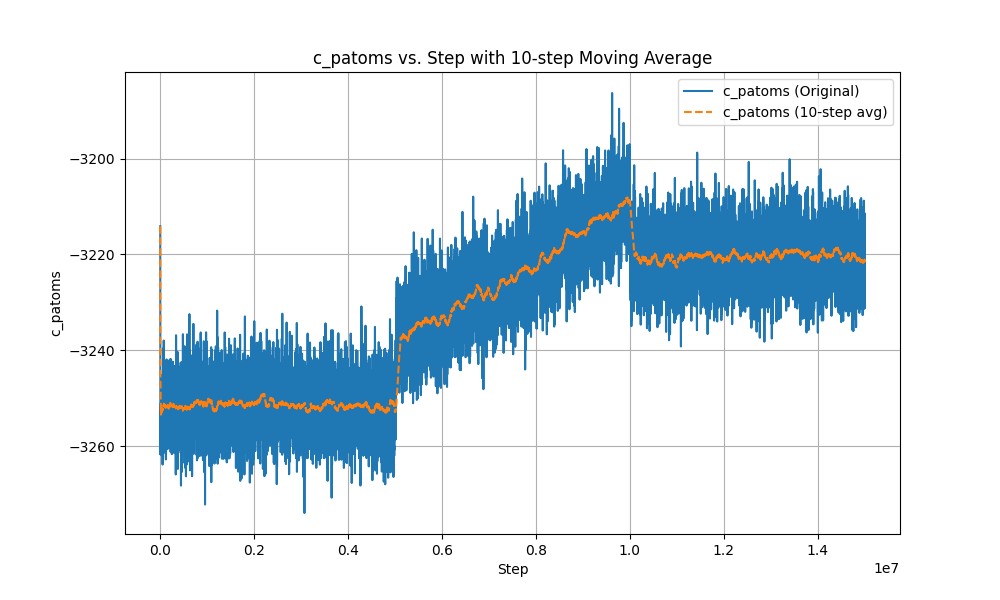
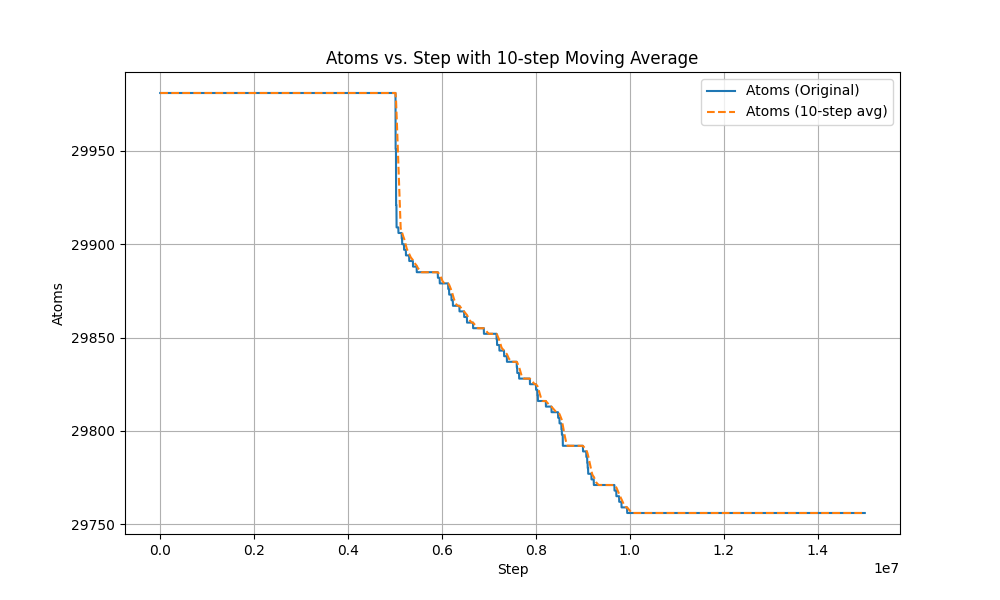
here is the atom-steps figure.
I am not clear about the reason for this contradiction, I think it is a LAMMPS bug for fix evaporate and here is my input:
units metal
dimension 3
boundary p p p
timestep 0.001
atom_style full
neighbor 3.0 bin
neigh_modify delay 0 every 1 check yes
read_data out.data
kspace_style pppm 1.0e-5
pair_style hybrid eam/fs lj/cut/coul/long 10
bond_style zero
angle_style zero
pair_coeff * * eam/fs Cu1.eam.fs NULL NULL Cu
pair_coeff 1 1 lj/cut/coul/long 0.00659558 3.1507 # o
pair_coeff 2 2 lj/cut/coul/long 0.0 0 # h
pair_coeff 1 2 lj/cut/coul/long 0.0 0 #o-h
pair_coeff 1 3 lj/cut/coul/long 0.052520151 2.720475282 #Cu-o
pair_coeff 2 3 lj/cut/coul/long 0 0 #Cu-h
bond_coeff 1 0.9574 # o*-h*
angle_coeff 1 104.52 # h*-o*-h*
region fixed block EDGE EDGE EDGE EDGE EDGE -10 units box
region evap_space block EDGE EDGE EDGE EDGE 90 EDGE
region elf_space block EDGE EDGE EDGE EDGE 1 80
group Water type 1 2
group fixed region fixed
set type 1 charge -0.834
set type 2 charge 0.417
compute peng all pe/atom
compute patoms Water reduce sum c_peng
compute keng all ke/atom
compute katoms Water reduce sum c_keng
variable wateng equal "c_patoms+c_katoms"
compute wtemp Water temp
compute wtempcom Water temp/com
thermo 1000
thermo_style custom step temp atoms v_wateng c_wtemp c_wtempcom c_patoms c_katoms etotal ke pe
fix freeze fixed setforce 0 0 0
fix 001 Water shake 0.0001 100 0 b 1 a 1
#fix evp Water evaporate 10000 30 evap_space 34255 molecule yes
fix fnvt Water nvt temp 295.35 295.35 0.1
run 5000000
fix evp Water evaporate 20000 30 evap_space 34255 molecule yes
dump 1 all atom 2000 dump_photo.xyz
run 5000000
unfix evp
run 5000000