Hi,
I am developing a workflow for constructing crosslinked models of thermoset polymers and predicting their thermomechanical properties. I’ve been able to use fix bond/react to produce fairly densely crosslinked inputs (70-85%) for several different epoxy-amine and epoxy-hydroxyl compositions, but I run into consistent errors on the annealing step.
Typical annealing workflows for these types of systems involve either NVT or NPT simulations at high temperature (600 K) to relax the network. A subsequent cooling simulation is then used to extract the density vs. temperature relationship and determine the glass transition temperature. When I try to replicate these literature workflows, I consistently run into ‘Bond atoms missing’ errors during the high temp relaxation.
I’ve tried a couple of what seemed to be the obvious fixes: reducing the timestep to 0.5fs, building neighbor lists every step, increasing the pair_style cutoffs, increasing comm_modify, and running the simulation on a single processor.
I’ve also tried several variations of the annealing workflow, including slowly ramping the temperature to 600K by setting the initial and final temps with fix npt. This approach does not generate errors, but the simulation volume often increases dramatically to produce an unreasonable final density.
The attached files reproduce both these types of errors and also include the equilibration and crosslinking inputs used to generate the thermoset model. The system (EPN1180 + Bisphenol-A) and NVT annealing workflow are based on the reference below [1].
Equilibration and Crosslinking Simulation:
Crosslinking.tar.gz (3.2 MB)
NVT Annealing method [1]
NVTAnnealing.tar.gz (545.3 KB)
NPT Ramped Annealing
RampedAnnealing.tar.gz (1.3 MB)
Plotted thermo outputs showing sharp dip in bond energy on second NPT ramp and bond length distribution for bonds formed & broken in fix bond/react.
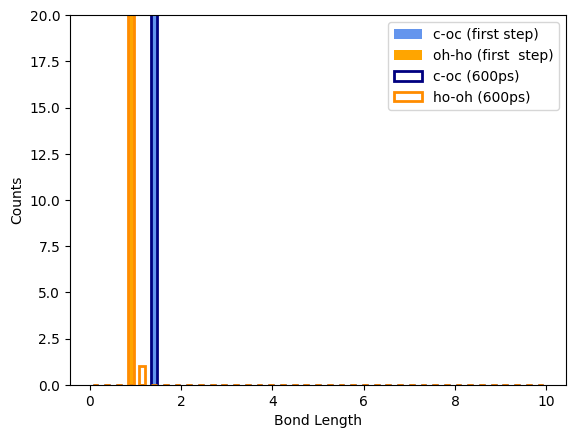
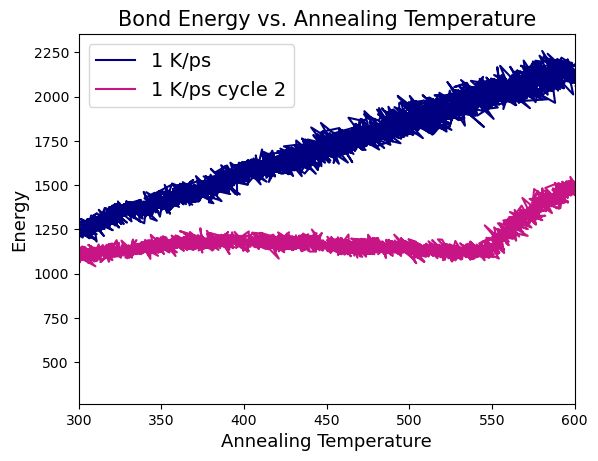
I’d appreciate any suggestions you might have on how to debug this issue. I’m still relatively new to LAMMPS and may have missed something obvious.
Thank you in advance for your help.
[1] Y. Shaorui & J. Qu. "Computing thermomechanical properties of crosslinked epoxy by molecular dynamics simulations."Polymer 53, 4806-4817 (2012). Redirecting