Hello everyone,
I’m running a simple SMD test case in LAMMPS, where a spring pulls a single particle. The relevant commands are:
create_atoms 1 single 0.03 0.03 0
fix pull tlattice smd cvel 100 0.1 tether 0.02 0.03 0.0 0
According to the documentation, for cvel mode, the spring’s equilibrium position is incremented linearly over time based on the specified velocity.
However, I’m encountering an issue when splitting the simulation into multiple runs:
run 3000
run 3000
or when using a restart:
run 3000
restart 3000 restart/restart*.bishear
Then in a new input script:
read_restart restart\\restart3000.bishear
fix pull tlattice smd cvel 100 0.1 tether 0.02 0.03 0.0 0
run 3000
In both cases, it seems the spring equilibrium position is re-initialized after each new fix smd command, and computed from the current position of the particle and the tether point, instead of continuing from the previous state. This causes the spring force to reset to zero, breaking the continuity of the force evolution.
In the output (see attached force plot), you can clearly see that after the second run or restart, the spring force drops back to zero instead of continuing smoothly.
My question:
- Is there a way to persist the internal state of
fix smd(specifically the current equilibrium position) across multipleruncommands or after arestart? - Or is there a recommended way to perform continuous SMD pulling across runs or simulations without force discontinuity?
Any suggestions or workarounds would be greatly appreciated.
Thanks in advance!
Here are the output curves of the spring force and equilibrium position.
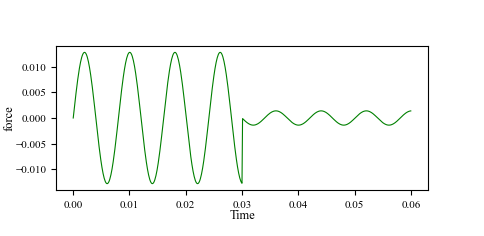
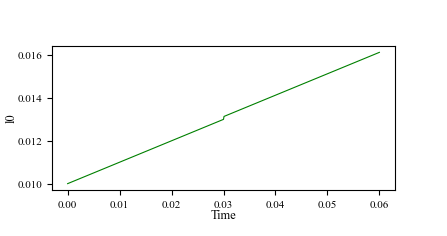
The following script can reproduce my question:
#! bin/bash
# file header
atom_style sphere
atom_modify map array
dimension 3
boundary p p f
newton off
comm_modify vel yes
units si
region reg block 0 0.06 0 0.06 -0.04 0.046 units box
create_box 1 reg
neighbor 0.001 bin
neigh_modify every 1 delay 0
#
pair_style granular
pair_coeff * * hertz 36630036630.03663 0.9 tangential mindlin 45248868778.28054 1.0 0.5 damping coeff_restitution
pair_style gran/hertz/history 37e9 45e9 1000 1000 0.5 1
pair_coeff * *
create_atoms 1 single 0.03 0.03 0
group tlattice id 1:1
set group tlattice diameter 0.005 density 2499.9999999999995
velocity tlattice zero linear
velocity tlattice zero angular
fix trlc tlattice aveforce NULL NULL NULL
fix pull tlattice smd cvel 100 0.1 tether 0.02 0.03 0.0 0
timestep 0.00001
compute 1 tlattice com
thermo_style custom step atoms f_trlc[1] c_1[1]
thermo 10
thermo_modify lost ignore norm no
shell mkdir post
shell mkdir restart
variable m_time equal time
fix integr all nve/sphere
fix ave_data all ave/time 1 1 10 v_m_time f_trlc[1] c_1[1] f_pull[1] f_pull[5] f_pull[6] file plate.txt title1 "" title2 "step time sf lx pullf l0 dx"
dump dmp all cfg 1000 post/dump*.cfg mass type xs ys zs id type x y z vx vy vz fx fy fz radius
restart 3000 restart/restart*.bishear
run 3000
run 3000