I want to simulate an ionic liquid using OPLS -AA force field. I made a data file which contains bond angle and dihedral information (not coefficients). I took torsional fourier coefficients V1 V2 V3 from a publication supporting doc but getting this error
ERROR: Incorrect args for dihedral coefficients (src/MOLECULE/dihedral_harmonic.cpp:251)
Last command: dihedral_coeff 1 -12.92 0 0 # C1-C2-N2-C3
I tried using dihedral_style ,harmonic ,opls ,fourier how can I fix this ?
Attaching the input script
IL.in (8.7 KB)
Can please someone point out where I am missing?
With the harmonic style that you are currently using in your input, shouln’d d be either 1 or -1 (you are using 0)? This is what the documentation suggests.
It sounds weird to randomly try different styles. I am sure that the style is imposed by the OPLS force field.
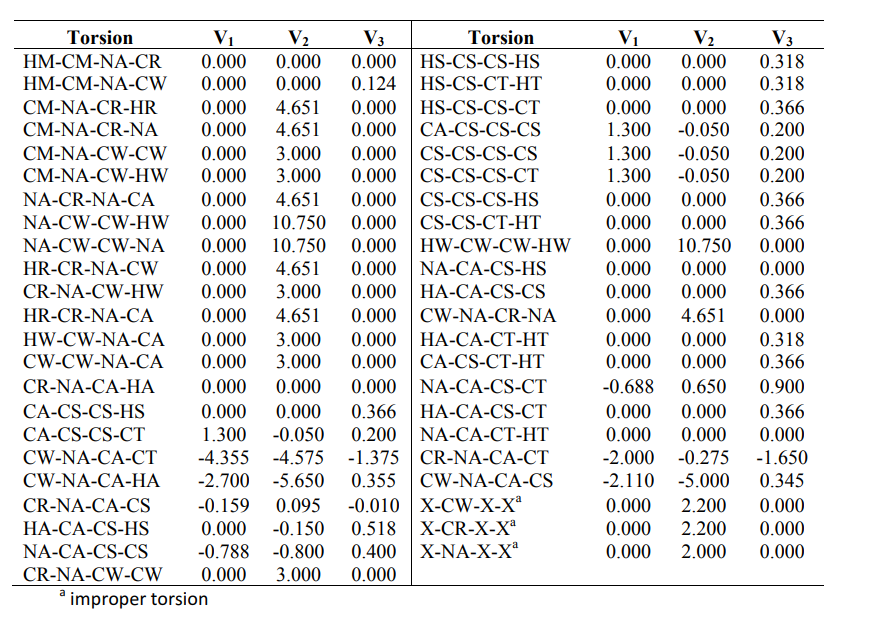
I tried to figure it out but OPLS style has 4 arguments and I have only V1 V2 V3 values
Well, may be this means that the 4 parameters should be chosen as 0? I am not sure of that though, I am no expert of OPLS force field.
I suppose you need to first know what is the form of the dihedral potential involved in your force field. Once you do it, you check the available ones within LAMMPS to find the one that duly matches. Then you learn the order in which you should input the coefficients (based on how many there are in the first place).
1 Like
What do you mean by form of dihedral potential ?
You have to look at the mathematical expression of the potential, and compare to the expressions given in the documentation of LAMMPS.
1 Like
I fixed it. I am having another error now missing bond atoms 18531 and 18544 on proc 0 at step 6824
explanation: atom ID 18531 is of particular atom type C and atom ID 18544 is of particular atom type H. I have EMIM cation residue type in my simulation system to which they belong. Only these two atoms of particular molecule ID are going out of the system. I tried changing bond constant, increasing epsilon values but did not help. Does someone have any insight on this?
Can you share your input script (preferably well formatted)?
PS: You are trying to make a classical simulation using a specific force field. Do you really think that changing the prescribed potential parameters is something you should be doing?
Please make an effort to follow the rules of the forum. You can’t just keep bringing new issues to an existing thread, which btw was already difficult to understand and was lacking many critical information (LAMMPS version, minimal input to reproduce the error, etc.).
Hi, OPLS-AA originally has 3 coefficients, and Fourier Function may include 4. Since OPLS-AA does not have the the fourth coefficient, then it should be zero in your input file where you define your forcefield.