Dear LAMMPS users,
I want to calculate thermal conductivity of single polymer strand by MP method. As provided by most strategies, I first fixed NVT, and then fix NVE and fix thermal /conductivity as shown in the code.
However, the total energy of the system continues going up and temperature does. I double checked everything without any solution. The pdb file during fix theraml/conductivity shows that the polymer chain tends to curved.
Can any one help me out ? thanks.
units real
atom_style full
dimension 3
boundary p p p
pair_style lj/cut/coul/long 10.0 10.0
pair_modify mix geometric
bond_style harmonic
angle_style harmonic
dihedral_style opls
special_bonds lj/coul 0.0 0.0 0.5
kspace_style pppm 1e-4
read_data PE.data
neighbor 2 bin
neigh_modify delay 0 check yes
variable fac equal 1.00
variable nSlab equal 40
variable Length equal 124
variable nSwap equal 1800
variable LenSlab equal {Length}*{fac}/${nSlab}
change_box all x scale ${fac} remap units box
minimize 1.0e-4 1.0e-6 100 1000
velocity all create 300 10
fix 1 all nvt temp 300.0 300.0 100.0 drag 0.2
fix 2 all momentum 1 linear 1 1 1 angular
timestep 0.1
velocity all create 300 12
run 1000
timestep 1
velocity all create 300 13
run 20000
unfix 1
unfix 2
fix NVE all nve
timestep 1
run 100000
reset_timestep 0
dump 1 all custom 1 dos.dump id type vx vy vz
dump 2 all custom 1000 bio_pdb.dump id type x y z
run 20000
undump 1
undump 2
reset_timestep 0
write_restart bioini.restart
compute ke2 all ke/atom
variable temp atom c_ke2*335.479
fix temp_profile all ave/spatial 1 {nSwap} {nSwap} x lower {LenSlab} v_temp &
file temp.profile units box
fix heat_swap all thermal/conductivity {nSwap} x {nSlab}
fix heatflux all ave/time {nSwap} 1 ${nSwap} f_heat_swap file heatSwap.data mode scalar
thermo_style custom step temp pe ke etotal f_heat_swap
thermo_modify lost warn
thermo ${nSwap}
dump 1 all custom 5000 bio.dump id type x y z
run 10000000
Part of the log file :
Step Temp PotEng KinEng TotEng heat_swa
8160000 304.54347 -677.46454 253.27236 -424.19218 2797.5843
8163200 295.2164 -668.67075 245.51554 -423.15521 2799.184
8166400 288.76888 -664.10069 240.15348 -423.9472 2799.7251
8169600 303.95069 -676.41776 252.77938 -423.63838 2800.18
8170000 304.90339 -676.88883 253.57169 -423.31714 2800.18
Loop time of 7.11138 on 16 procs for 10000 steps with 280 atoms
Memory usage per processor = 13.9479 Mbytes
Step Temp PotEng KinEng TotEng heat_swa
16260000 328.14823 -679.42943 272.90317 -406.52626 5722.6631
16262400 311.54201 -664.91377 259.09267 -405.8211 5722.7674
16265600 310.30801 -664.84287 258.06642 -406.77645 5724.855
16268800 308.99398 -663.63225 256.97361 -406.65864 5725.9905
16270000 308.2574 -662.20872 256.36103 -405.84769 5725.9905
Loop time of 7.48647 on 16 procs for 10000 steps with 280 atoms
Step Temp PotEng KinEng TotEng heat_swa
68050000 360.45114 -608.16249 299.76775 -308.39474 26507.826
68051200 335.11148 -585.93862 278.69413 -307.2445 26509.625
68054400 377.2177 -622.01881 313.71159 -308.30721 26510.347
68057600 364.54733 -611.5104 303.17433 -308.33607 26511.862
68060000 372.17516 -618.56509 309.51799 -309.04711 26511.862
Loop time of 7.69513 on 16 procs for 10000 steps with 280 atoms
Step Temp PotEng KinEng TotEng heat_swa
104270000 397.69803 -568.33525 330.74397 -237.59128 43261.797
104272000 421.48862 -588.20212 350.52933 -237.67279 43263.375
104275200 411.4236 -578.57889 342.15879 -236.4201 43266.259
104278400 424.79958 -590.14994 353.28288 -236.86706 43265.63
104280000 430.50965 -596.10339 358.03163 -238.07176 43265.63
Loop time of 7.40201 on 16 procs for 10000 steps with 280 atoms
pdb file

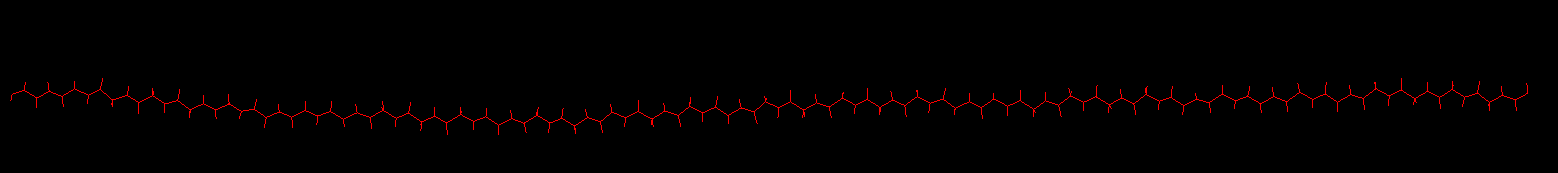
Thanks for help.
John Zhang

