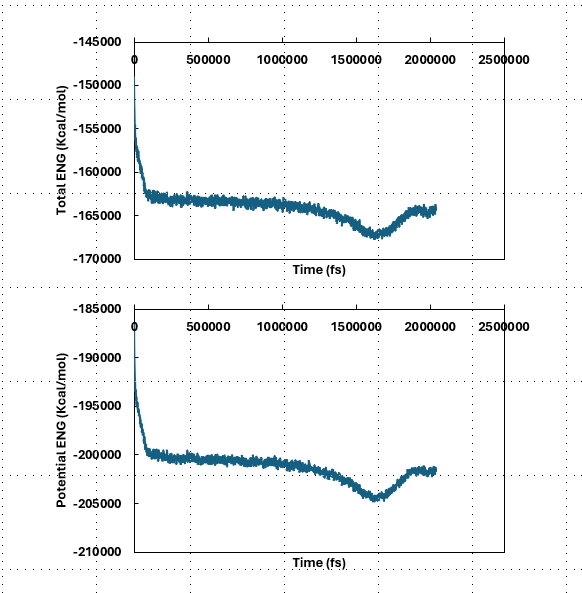
I have a system composed of 20 polymer molecules and 2200 water molecules (21060 atoms in total). I am using NPT at 600 K to equilibrate my system for 3000 ps. Starting after a few pico seconds, the total energy of system shows a plateaued phase, which continues until 1000 ps. But after this time, the equilibration is disrupted and the energy starts to become unstable. At this stage, the system also starts to expand again (density goes from 1.6 to 0.2 g/cm^3). I have checked the kinetic and potential energy of the system and the same trend can be seen for the potential energy, while kinetic profile stays stable. I have tried several approaches, such as increasing the temperature of only polymer molecules to 600 K and keeping the temperature of water molecules at 300 K. However, this does not work, either. I was wondering if anyone can help me to figure out the reason behind such behavior in my system.
I should mention that for my other systems with the same polymer molecules, but less number of water molecules, this has not happened and system seems to be working fine. I should also mention that the system has no charge, as well.
You need to describe your equilibration protocol more quantitatively, e.g. by providing a stripped-down input file showing the force field used, the coupling constants, etc.
Your simulation runs for 2.5 ns, contains water, and is run at 600 K. It does not surprise me that you observe a metastable state that suddenly turns to vapour. What would you expect to happen instead?
Dear @hothello, thank you for your response. I attached the input file here. Actually, I tried to impose temperature only on the polymer to eliminate the impact of high temperature on water expansion/evaporation. However, again the system was expanded and the energy was fluctuating after a while.
In fact, I tried imposing 600 K temperature both on all system and only on polymer particles.
echo both
#-------------------------------------------------------------------------------
# Stage 2.1: Initialize LAMMPS run for 3-d periodic
#-------------------------------------------------------------------------------
units real
boundary p p p
atom_style full
restart 200 restart
pair_style lj/class2/coul/long 10
pair_modify mix sixthpower
pair_modify tail yes
bond_style class2
angle_style class2
dihedral_style class2
improper_style class2
special_bonds lj 0.0 0.0 1.0 coul 0.0 0.0 1.0
box tilt large
#read_data structure.dat
read_restart structure.dat
include parameters.dat
neighbor 2.0 bin
neigh_modify delay 0 every 1 check yes
kspace_style ewald 1.e-5
#velocity all create 300.0 497 dist gaussian
#minimize 1.0e-6 1.0e-6 5000 5000
#group of chains-------------------------------------------------------------------------------
group chain1 id 1:682
group chain2 id 683:1364
group chain3 id 1365:2046
group chain4 id 2047:2728
group chain5 id 2729:3410
group chain6 id 3411:4092
group chain7 id 4093:4774
group chain8 id 4775:5456
group chain9 id 5457:6139
group chain10 id 6140:6821
group chain11 id 6822:7502
group chain12 id 7503:8184
group chain13 id 8185:8866
group chain14 id 8867:9548
group chain15 id 9549:10230
group chain16 id 10231:10912
group chain17 id 10913:11594
group chain18 id 11595:12276
group chain19 id 12277:12958
group chain20 id 12959:13640
group Polymer id 1:13640
group Fluid subtract all Polymer
#Exports ......................................................................................................................................
timestep 1
thermo 1000
thermo_style custom step cpu temp pe ke epair ecoul etotal density lx ly lz
dump 1 all xyz 3000 System.xyz
#reset timestep
#reset_timestep 0
# Fix shake is needed for water in pcff+
fix shaken all shake .0001 20 50000 a 12 b 7
#nvt run
#fix 1 all nvt temp 600.0 600.0 50.0
#run 1000000 upto
#undump 1
#write_restart restart-1.equil
#unfix 1
#..............................................................................................................................................
#reset timestep
#reset_timestep 0
#run at high temperature for 3000 ps
fix 2 Polymer npt temp 600 600 50 iso 1 1 1000 drag 0 mtk yes nreset 10000
fix 3 Fluid nvt temp 300.0 300.0 50.0
run 3000000 upto
#write_restart restart-1.equil
unfix 2
unfix 3
undump 1
If you don’t want the system to expand, then why do you keep running with fix npt?
Why not just stop the npt run way before the expansion starts and continue with fix nvt?
You probably should discuss about general strategies with a more experienced person locally.
It is not the job of the forum to teach people how to do research, but rather discuss LAMMPS, and LAMMPS is just doing what you are asking it to do, so there is nothing to discuss.