Dear LAMMPS users and developers,
I carried out SPC/E-water simulations using Wolf-method (wolf.lmp) and Ewald summation (ewald.lmp) for electrostatics, respectively. I found that the total energy as calculated with the Wolf method is about a factor 8 higher as compared to Ewald summation. However the kinetic energy is approximately the same. Here is the LAMMPS output:
Wolf-Method:
Wolf summation is only comparable to Ewald summation with carefully
chosen parameters, i.e. the alpha and the cutoff. You might want to
experiment more with these parameters.
Also in your ewald.lmp I don't see a pair_style. That could be a
major source of difference.
Ray
Hi Ray,
thanks for the quick reply! The parameters for the Wolf simulation are chosen carefully, I think, as I took them from a recent publication on the exact same system. With regard to the pair_style and the Ewald-run, the information should already be contained in the binary restart file. According to the documentation, the only thing which requires to be set again is “fix shake” and “kspace ewald”. The situation does not change, if I melt the crystal with Ewald/Wolf-summation and continue with a short NVE-run in the same input file, respectively. Therefore I would rather suspect a different problem.
However, I tried to be more careful with the pppm binary restart file and subsequent Wolf-summation, as there could be something set/unset due to the restart. Therefore, I carried out another NVT Wolf-simulation (melt_wolf.lmp) to compare with the previous NVT PPPM-simulation (melt.lmp). The energies are again an order of magnitude off.
Here is the output from that 10 ps NVT-run (generated with melt_wolf.lmp):
Wolf summation generates comparable results to Ewald if 1) Wolf
parameters are carefully chosen, 2) input structures are the same, and
3) potential parameters and other setups are the same.
There must be something from 1-3 that is causing the difference. You
might also try not using fix shake and other non-necessary commands to
see if you can get the same results.
Ray
Hi Ray,
ad 1) the parameters are carefully chosen.
ad 2) the input file spce_2592.dat was the same; both runs started with the identical file equi.res obtained from melt.lmp in a NVT simulation
ad 3) LJ-parameters are the same
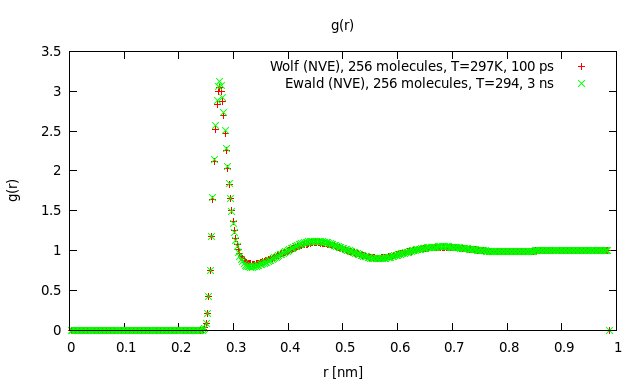
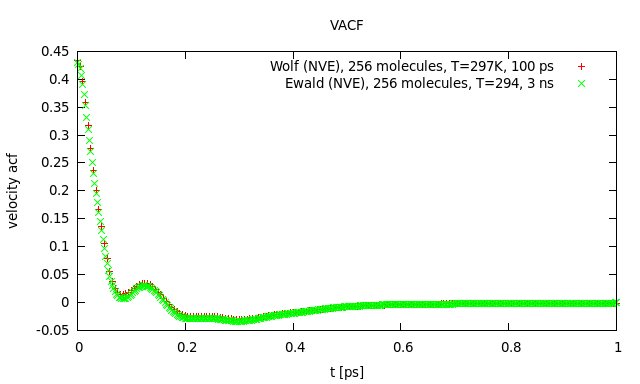
I launched another short simulation (100 ps) using the Wolf-script for 256 molecules. I dumped the coordinates and velocities to a file every 5 time steps and did some post-processing to get g® and the velocity autocorrelation function. I then compared the results with previous results obtained by Ewald summation (please find gr_comp.jpg and acf_comp.jpg attached). As you can see, they agree very well. The slight deviation in the maximum of g® is due to the temperature difference of 3 K. This does not look like I got the input parameters terribly wrong. The static dielectric constant of the short run (100 ps) was 83 (literature value for SPC/E is 71). That seems reasonable, as you need at least 1 ns to get close to the literature value because the dipole moment relaxes quite slowly.
Anyway, something is wrong here, I agree.
Best, Peter
I think you should verify this on your own. Publications can have typo’s.
Were the first step energies and forces from Wolf and Ewald the same?
If not, then something from 1-3 MUST be wrong.
Ray
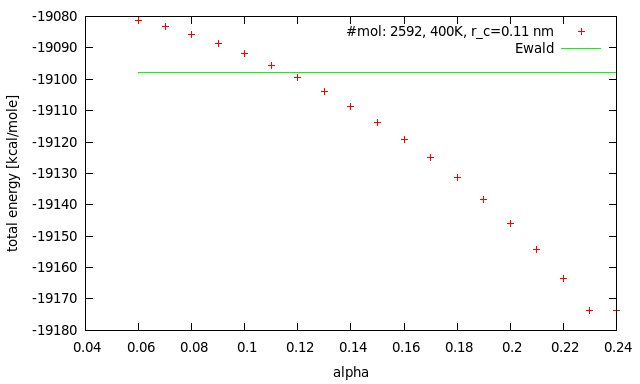
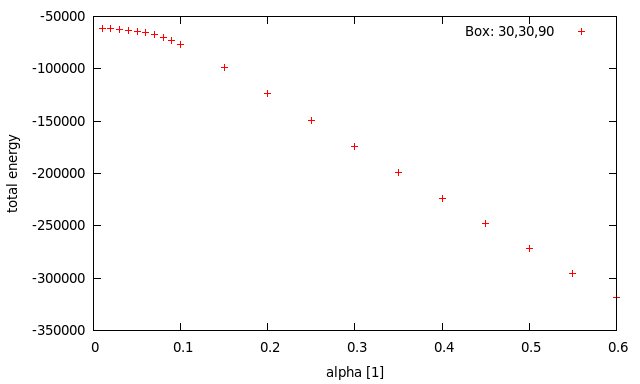
Please find attached a plot of the total energy as a function of alpha, the damping factor of the Wolf method for a fixed cut-off of 11 Angstom. In (J. Chem. Phys., Vol. 110, No. 17, pp 8254-8282) Wolf et al. carried out simulations of MgO and used a value of alpha=1.2/a0=0.28 for their simulation (a0=4.2). In my examples I used 7.2/L=0.24 (L=30). The LAMMPS documentation suggests a value of 0.2. To be more specific, what you see in the plot is the total energy after 50 NVE steps with the Wolf-method starting with a previously equilibrated binary restart file (see melt.lmp). The energy is conserved for both runs respectively. The input file “equi.res” was equilibrated before with pppm. The whole system is charge neutral. There is a plateau for very low values of alpha, but not where I expect it to be. Just as a reference, the last energy of the pppm NVT run was ~ -19000.
@Stan:
thanks a lot. The system is charge neutral and I am simulating bulk water. I will send you an archive wolf.tar (4.3 MByte) in a separate email. as tp avoid approval. That contains all the input scripts from above as well as a file README and the lammps output. Please note, that in the melt*.lmp files in the archive I just used 1.5 ps to melt the crystal, just for demonstration, whereas in my initial posts it was 10 ps.
@Ray:
If they were roughly the same, there would be no need for the post. Again, I agree that there must be something wrong. The question remains, what it is. Most likely my scripts, but I cannot find a mistake.
Here is the per time step output.
Ewald:
I found the source of difference, but I don't know what is the reason
yet. It seems that bond_style triggers a significant change to
ecoul, which is the Coulombic pairwise energy.
Below are the results of my test using the scripts attached. E1 and
W1 stands for pure atomic Ewald and Wolf (with bonds and angles
sections taken out from the data file), and E2 and W2 are Ewald and
Wolf with bond_styles (angle section taken out).
You can see that Ewald and Wolf are comparable for pure atomic, but
they differ significantly when bonds are used, particularly the
E_coul.
in.ewald (1.04 KB)
in.wolf (1.13 KB)
ray,
this would likely be related to exclusions leading to entries being
removed from the neighbor list. if i am right, you should achieve the
same effect without removing bonds, but setting:
special_bonds coul 1.0e-100 1.0e-100 1.0
axel.
Thanks, Axel! I forgot about the neighbor list exclusions!
Peter, all you need to do is to add the following to your scripts and
you will get the same results from Ewald and Wolf (provided that
bullets 1-3 satisfy).
special_bonds coul 1.0 1.0 1.0
Ray
Thanks, Axel! I forgot about the neighbor list exclusions!
Peter, all you need to do is to add the following to your scripts and
you will get the same results from Ewald and Wolf (provided that
bullets 1-3 satisfy).
special_bonds coul 1.0 1.0 1.0
sorry, i don't think that this is going to be correct. both
trajectories will be equally wrong, at least when run w/o shake.
please compare this with my suggestion of using almost zero for 1-2
and 1-3 terms. after all, you need to make those terms vanish.
after looking at the coul/wolf code for a bit, i am at a bit of a
loss, why tricking the neighbor list build into including pairs, that
do not contribute, would make such a big difference, though.
axel.
I think this has to do with neighbor.cpp, line 323:
if (force->kspace) special_flag[1] = special_flag[2] = special_flag[3] = 2;
This line is true for the ewald case but not for wolf summation, which leads to a different behavior.
In other words, the default setting for the special_bonds coul is different if a kspace is defined or not. That is why explicitly setting the special_bonds coul flag makes the two energies agree, because now the have the same settings (before they did not).
Stan
Thanks, Stan and Axel.
Does this mean that Wolf needs an additional line of code to set the
special_flags?
Ray
Thanks, Stan and Axel.
Does this mean that Wolf needs an additional line of code to set the
special_flags?
after staring at the code some more, i think i am beginning to see
what is going on and the answer to that question would be: yes.
the problem is: 1-2, 1-3, 1-4 exclusions apply to the *full* coulomb
interaction (which is typically stored in "prefactor"),
however, both coul/long and coul/wolf compute a *damped* coulomb
interaction which incorporates components from long-range coulomb that
must not be excluded. so to completely drop the excluded interaction
(due to completely dropping the pair from the neighbor list) would be
an error. thus when using coul/wolf in any form, the same rules as for
including kspace apply and something like the following change would
be needed. provided that my analysis is correct, that is.
[[email protected]... src]$ git diff neighbor.cpp
diff --git a/src/neighbor.cpp b/src/neighbor.cpp
index 10f7ce0..133b184 100644
--- a/src/neighbor.cpp
+++ b/src/neighbor.cpp
@@ -320,7 +320,8 @@ void Neighbor::init()
special_flag[3] = 1;
else special_flag[3] = 2;
- if (force->kspace) special_flag[1] = special_flag[2] = special_flag[3] = 2;
+ if (force->kspace || force->pair_match("coul/wolf",0))
+ special_flag[1] = special_flag[2] = special_flag[3] = 2;
// maxwt = max multiplicative factor on atom indices stored in neigh list
Yeah that is the conclusion that I am coming to as well. This would also apply to the pair_style cou/dsf (a modified version of Wolf) and related suffix styles. Perhaps adding another flag to these pair_style would be good.
Stan
Another issue about these pair_styles is that they include a single() function but that doesn’t include the self-energy portion, which would makes results from say compute group_group wrong. Maybe we should disable the single() function.
Stan
Yeah that is the conclusion that I am coming to as well. This would also
apply to the pair_style cou/dsf (a modified version of Wolf) and related
suffix styles. Perhaps adding another flag to these pair_style would be
good.
using the "inexact" version of Force::pair_match() will catch all of
those cases (note the "0" instead of "1"):
diff --git a/src/neighbor.cpp b/src/neighbor.cpp
index 10f7ce0..0fe4672 100644
--- a/src/neighbor.cpp
+++ b/src/neighbor.cpp
@@ -320,7 +320,9 @@ void Neighbor::init()
special_flag[3] = 1;
else special_flag[3] = 2;
- if (force->kspace) special_flag[1] = special_flag[2] = special_flag[3] = 2;
+ if (force->kspace || force->pair_match("coul/wolf",0)
+ || force->pair_match("coul/dsf",0))
+ special_flag[1] = special_flag[2] = special_flag[3] = 2;
// maxwt = max multiplicative factor on atom indices stored in neigh list
however, this kind of change is indeed a bit difficult to track down.
at the very least some kind of comment near the constructor explaining
that an update of neighbor.cpp may be needed for derived classes
should be added. but something similar to ewaldflag, pppmflag or
tip4pflag would probably be a cleaner solution.
axel.
Dear all,
thanks a lot for investing so much time in clarifying this problem. I am impressed by how active this forum is.
@ Stan: