I’m trying to trace down the source of some infrequent occurrences of the error: Out of range atoms - cannot compute PPPM (src/KSPACE/pppm.cpp:1887)
The error occurs infrequently, often several nanoseconds into an otherwise stable run, or not at all. I have tried increasing neighbor list rebuilding by setting “neigh_modify delay 1”, which reduced occurrences of the error, but did not eliminate it.
The solution does seem to be clearly solvable with either more frequent neighbor list rebuilding or a larger skin distance. If I try and output a dump or restart on the timestep immediately before a timestep where the error is known to occur, the simulation will run fine with no error, presumably because the writing the dump forced the neighbor list to rebuild. Forces and velocities seem normal in the timesteps preceding the error.
I saw in a previous discussion on the email list that returning a more specific error, like the atom ID of the out-of-range atom, is not generally desirable because of the potential slowdown it could incur in the code. However I’m wondering if the developers have any suggestions on how I could temporarily add this to the code so I can trace the specific source of this error in my models more quickly and with more confidence.
For reference, I’m running June 2, 2022 LAMMPS, real units, a typical bio-type force field (lj/cut/coul/cut, harmonic bonds, angles, etc.).
What happens, if you reduce the delay to 0? What about reducing the timestep?
The error message already tells you the file and line where the error function is called. So you need to look at the preceding block of code if you want to add any atom specific tracking code.
Thank you for the suggestions Axel. With neigh_delay 10 it errors in ~14 ps. With neigh_delay 1 or 0 it errors at the exact same timestep at around 6.8 ns. I have a restart file ~60 ps preceding the error, I’ll see if I can reproduce it quicker.
Below are some timings and neighbor list statistics with various changes in neigh_delay, skin distance, and time step from 10 ps benchmarking:
neigh_delay 10, neigh_skin 2.0 A, time step 1.0 fs, (runtime 1.00x)
Neighbor list builds = 1000
Dangerous builds = 1000
neigh_delay 0, neigh_skin 2.0 A, time step 1.0 fs, (runtime 1.04x)
Neighbor list builds = 1229
Dangerous builds = 0
neigh_delay 10, neigh_skin 3.0 A, time step 1.0 fs, (runtime 1.00x)
Neighbor list builds = 617
Dangerous builds = 0
neigh_delay 10, neigh_skin 2.0 A, time step 0.5 fs, (runtime 1.84x)
Neighbor list builds = 1252
Dangerous builds = 0
Based on these results and the longer 6+ns run, increasing the skin distance is required and/or decreasing the time step with time step being the more expensive of the two options.
I’ll take a look at modifying pppm.cpp file and see if that turns anything up. Any additional suggestions on how I can visualize/flag atoms moving more than the skin distance are welcome.
The question that comes to my mind at this point is whether a time step of 1.0 fs is at all suitable for your simulation. Are you using fix shake to constrain all bonds involving hydrogen atoms? If not, reducing the time step would be the physically correct approach.
In your situation, I would at this point start looking at energy conservation with the 1.0 fs time step. It may be rather bad and the “Out of range atoms” situation is just a symptom caused by atoms moving too far for the steepness of their potential relative to their mass.
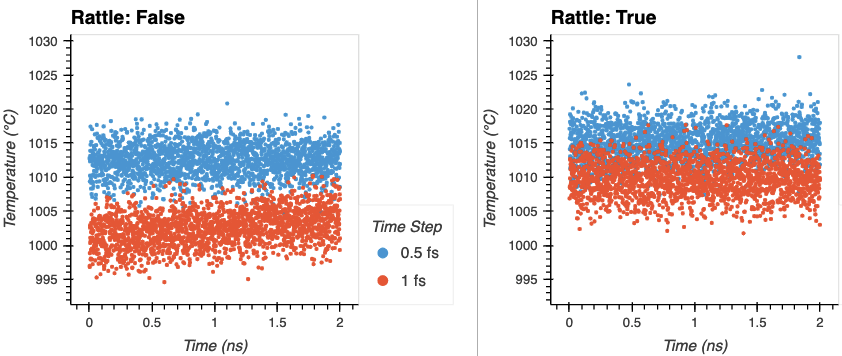
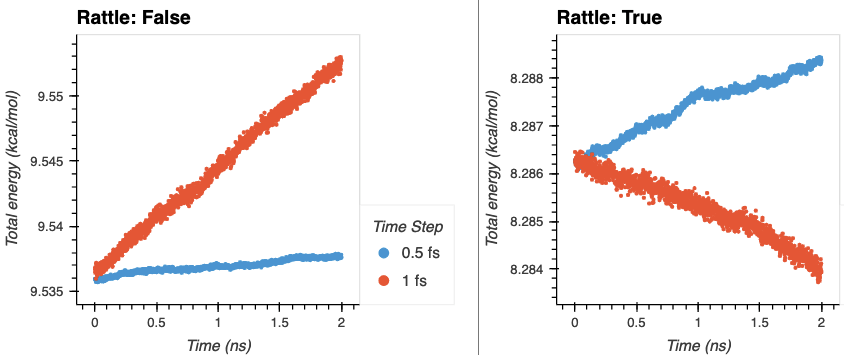
Agreed. I’ve been running without shake thus far. Energy conservation and temperature drift results are below for various combinations of time step and fixing hydrogen bonds over a 2 ns NVE run (with the appropriately aggressive neighbor delay and skin to prevent dangerous builds). There is little drift when using rattle. The temperature drift doesn’t seem too bad for the 1 fs time step without rattle to my eye (1.5 K/ns) for a simulation that will ultimately be controlled with NPT, but perhaps this is just because the low mass of the hydrogens mask their impact on the total temperature.
This is using Open Force Field, which I expect to behave similar to AMBER and OPLS. My understanding was that 1 fs was typical for flexible hydrogens, and 2 fs for fixed hydrogens, but I haven’t done a lot with bio force fields before this. As always, input is welcome.
My experience (mostly from water models) is that 0.25fs is a safe choice for flexible models (i.e. no SHAKE), 0.5fs is aggressive. It works better on larger molecules because the hydrogens are more buried. 1fs I would consider too aggressive. That is a safe choice with SHAKE on hydrogens and 2fs an aggressive choice. Way back when, when CPU time was much more scarce and hard to come by than today we occasionally used 2.5fs with SHAKE when we knew that we didn’t do analysis of the dynamics.
If you don’t want to use constraints, you can consider r-RESPA and do computation of bonds/angles more frequent than the rest. That can improve performance at the limit of strong scaling, because you can often do the kspace force computation only on every other pair style step.
I recently had a situation where I had to rigidify an angle, not just bond lengths. Sometimes you can run diagnostics using compute bond/local and compute angle/local and see what’s getting distorted.
This also has the advantage of telling you if multiple molecules in your system are doing poorly – the “print tag of lost atom” approach can only tell you the last atom lost unless you put a bit of machinery in (i.e. make a vector of lost particles and pushback).
Thank you @akohlmey and @srtee for the suggestions. r-RESPA seems to help, running with 1 fs long-range and 0.5 fs bonds/angles/etc is stable. It’s still strange to me that I have to run with around half the typical time step for this style of forcefield (typical is 1 fs flexible hydrogen, 2 fs w/shake, 4 fs w/shake and r-RESPA). It could be a force field issue, I know Open Force Field is pretty new still, or just my expectations for my system. Thanks again for the assistance.