Hi,
When I installed according to the steps on QUIP’s official website (exactly the same), I encounter the following problems during simulation. Following is the in file I adopted.
xml file is from Deringer’s model(aC_GAP_data_main\training\full_das\carbon.xml)
units metal
dimension 3
atom_style atomic
boundary p p p
# create region including Fixed layer, Constant temperature layer, Newton layer
# whole system
region box block -0.446 31.7 -0.446 31.7 -38 80 units box
create_box 1 box
lattice diamond 3.56683
# for cutting1 tool
region bulkn block 0 31.4 0 31.4 -34 -10 units box
# create atoms
create_atoms 1 region bulkn units box
# set mass for all atoms
mass 1 12.0110
neighbor 2.0 bin #2.0nm
neigh_modify every 1 delay 5 check yes
# add interacions
pair_style quip
pair_coeff * * carbon.xml "IP GAP label=GAP_2016_10_18_60_23_11_22_108" 1
timestep 0.0005
fix 1 all nve
run 10
Problem 1:
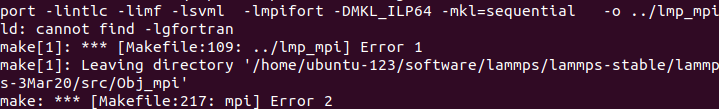
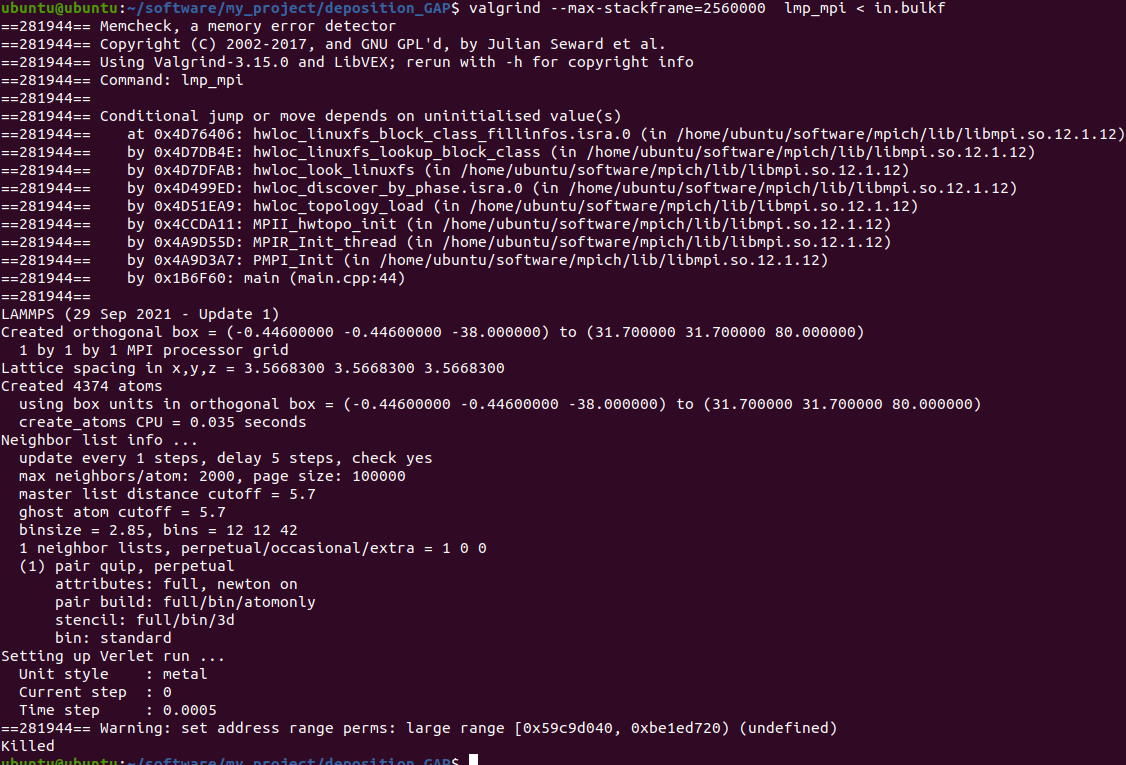
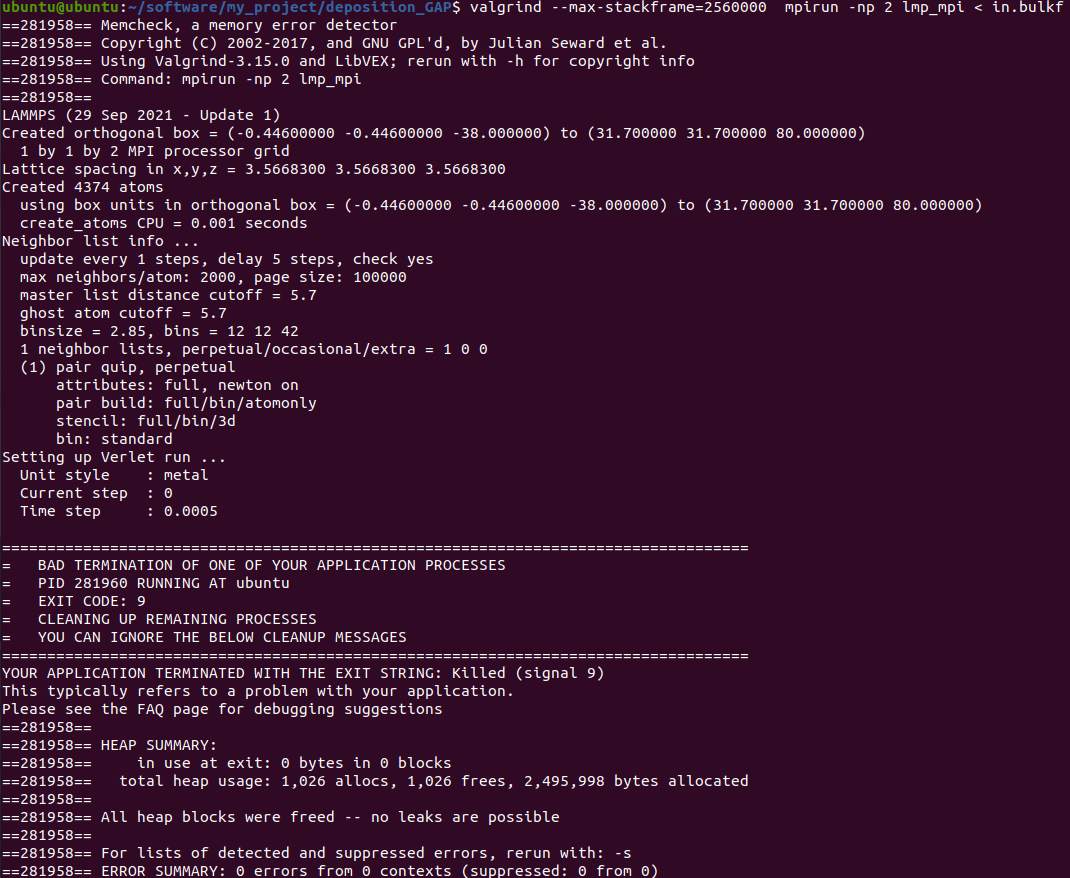
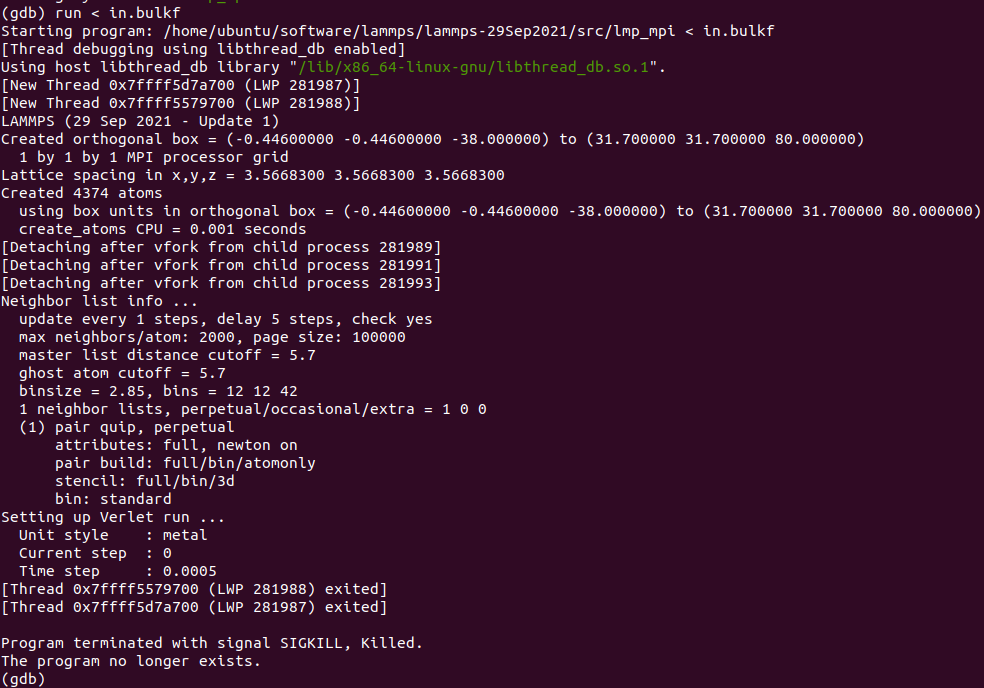
When I run with these files, something is wrong. The first figure shows the error message of single-core operation, the second figure shows the error message of multi-core operation, and the third figure shows single-core operation detected by Valgrind.
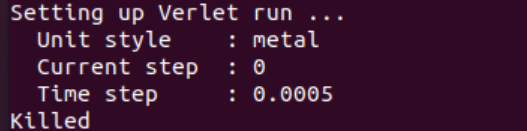
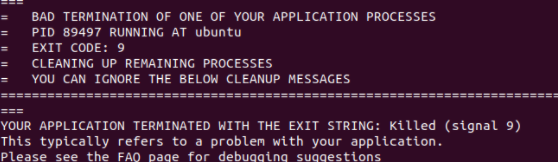
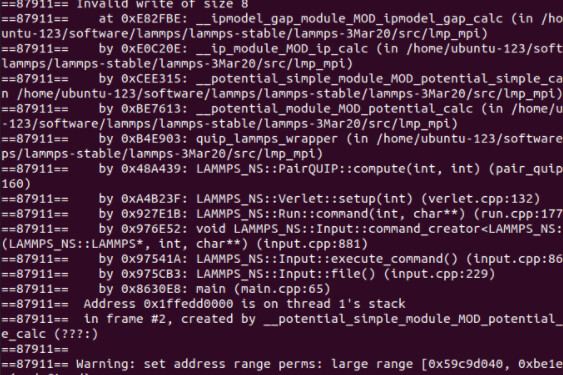
The correctness of in file has been verified much times with xml file in LAMMPS’s examples and CEDIP. Further testing revealed that the offending code was in line 9 of the in file. When I replaced this line with “region box block -0.446 10 -0.446 10 -38 80 units box” , everything is OK.
Problem 2:
The simulation speed is too slow to satisfy my requirements.
Each step takes three to five minutes(About 4000 atoms) and four cores require as much computation time as a single core. I don’t know what’s wrong with my QUIP installation or Parallel computing.
I test some xml files and found that all parallel operations did not improve the speed, but my installation steps are exactly the same as the official website. What might be the problem?
Best regards,
LZQ