Dear all,
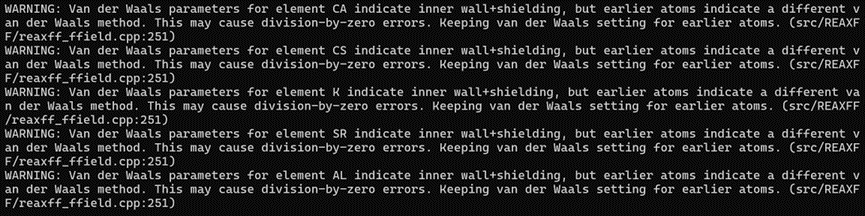
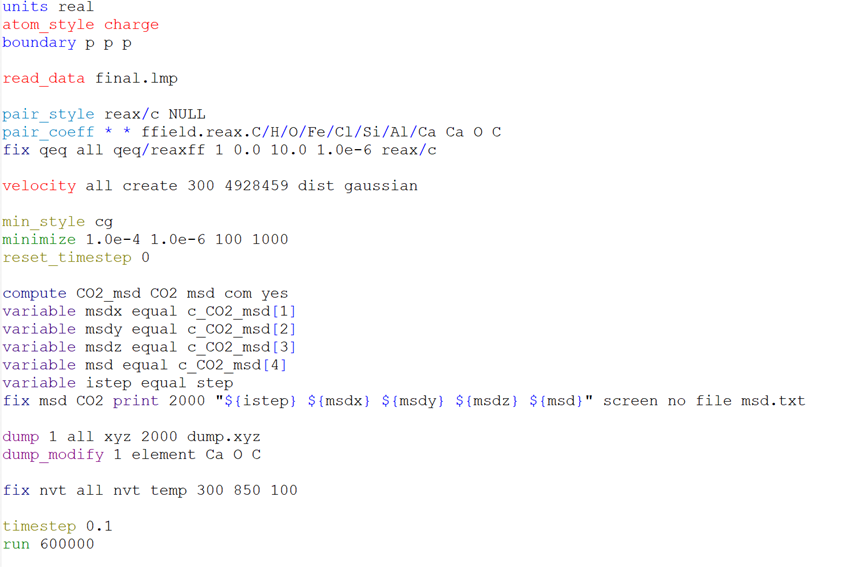
I’m new to using Lammps. I was using lammps to simulate the work of CO2 adsorption in CaO pipes and then calculate the msd of CO2, but I ran into some issues like the problem with the ReaxFF force field, I could only find this ReaxFF force field file for C/H/O/Fe/Cl/Si/Al/Ca, but this happened
I don’t know how to modify it, and at the same time I didn’t find the right QEq parameters when balancing the potential.
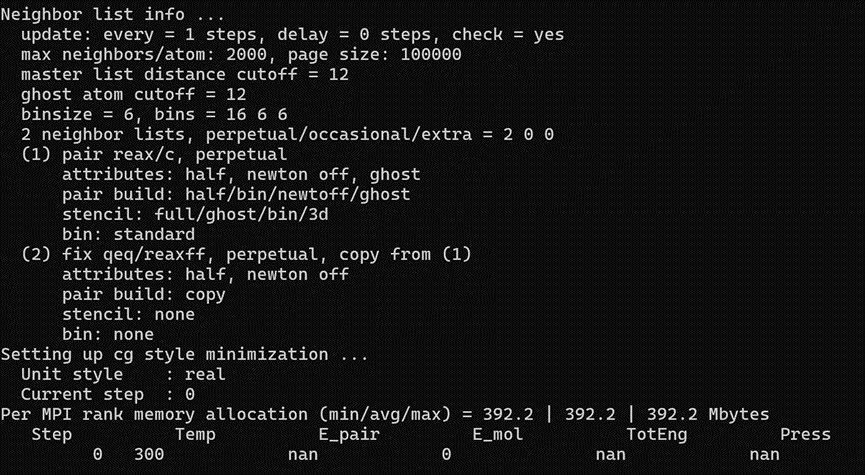
In step 0, the following happens:
Hi @dhly1,
Please have a look at this post to help you format your question so that it helps other reading it and providing meaningful answers.
ReaxFF is a complicated forcefield to use. I wouldn’t recommend starting with this one if you are new to LAMMPS. You should also look for local guidance to explain you the format and the forcefield if any other user near you already did such simulations. Besides, if you have no reaction or molecule dissociation in your system, there is no reason to use a reactive model, especially if you want to compute long time properties.
Where? It is nearly pointless to provide the name of the file without either the source or the file itself. The warnings (not errors) you show are likely to be caused by bad formatting or parameters declaration.
Besides the warning issue with the potential, that can also stem from your initial configuration, there are other issues with your script. You did not provide which LAMMPS version you are using, and the reax/c naming has been discontinued for 3 years. Consider updating your LAMMPS version.