Dear lammps users.
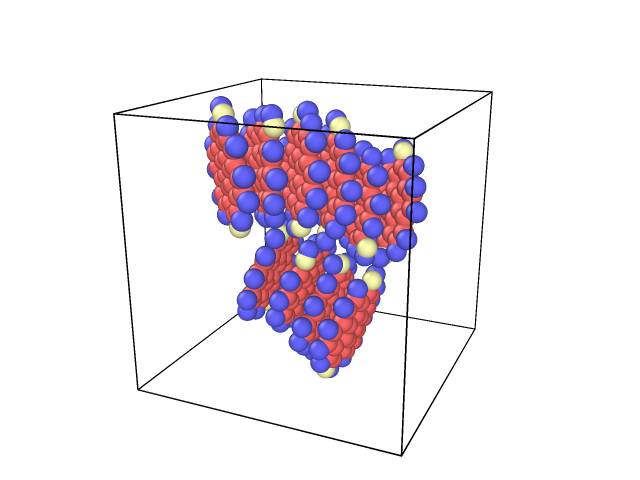
I have ten molecules in my system (each molecule is coronene with two hydroxyl attached to it) and Fig.1 is visualization of my system (blue, red and white balls are hydrogen, carbon and oxgen atoms). Firstly it was optimized using Material studios. Secondly, the structure was exported to lammps and was equilibrated at room temperature (20 ps), and then I heated it to 3000 K with heating rate 20 K/ps, two molecules are treated as a pair and constrained at desired region by using fix drag command when it was heated, finally the system were allowed to react at assigned temperate without fix drag command. The lammps and Reaxff forcefield (pair_style reax/c) were used for whole processes except optimization process. The simulation is normal until heating stage. But I got this error message during the reacting stage after some steps.
Error message
step1434819-hbondchk failed: H=1332 end(H)=909325113 str(H+1)=1575
My version is 10 Aug 2015, and my system is Fedora. Attachment1 and attachment2 are input for heating and reacting. What does it mean? What causes this error?
Sincerely
Fan Li
input for heating.docx (14.7 KB)
input for reacting.docx (14.3 KB)
hbondchk error has been widely discussed on the mailing list. Please do a little search on this topic. In short, this error usually occurs when you have bad dynamics or very rapid changes in your system. You can try increase safezone and mincap numbers. If you wish, you can attache your restart file and I will take a look.
Ray
Hi Ray
Thanks for answering my question.
The calculation is still running after increasing safezone and mincap numbers.
The attachments (coro-oh.reatart.1396000) are restartfile where the data was read at the beginning of the reacting stage. While another one (coro-oh.restart.1434000) was the last restartfile before calculation stopping.
I have looked at them. And I have several questions
(1)The restartfile have additional three columns comparing the data file which was used for equilibrium stage. What dose them mean?
(2)In coro-oh.reatart.1396000 file, most all of the values in these columns are 0, while a lot of 1 and -1 appears in coro-oh.restart.1434000 file. What 1 and -1 represent? Are they related to the stop of my calculation?
(3)Would you tell me how to find problems from the restart file? Shoud I check the coordinates of the atoms? If so, there are too many to check.
coro-oh.reatart.1396000.doc (103 KB)
coro-oh.restart.1434000.doc (103 KB)
Hi Ray
Thanks for answering my question.
The calculation is still running after increasing safezone and mincap
numbers.
The attachments (coro-oh.reatart.1396000) are restartfile where the data was
read at the beginning of the reacting stage. While another one
(coro-oh.restart.1434000) was the last restartfile before calculation
stopping.
please do not attach text file data as word documents. you are making
it needless complicated to save them. just attach them as plain text
directly.
I have looked at them. And I have several questions
(1)The restartfile have additional three columns comparing the data file
which was used for equilibrium stage. What dose them mean?
those are not restart files, but data files. restart files are not
human readable binary files.
those extra columns are image flags (assumed to be 0, if not present).
please see the documentation of the read_data command for a detailed
description of the data file format.
(2)In coro-oh.reatart.1396000 file, most all of the values in these columns
are 0, while a lot of 1 and -1 appears in coro-oh.restart.1434000 file. What
1 and -1 represent?
please see the previous answer.
Are they related to the stop of my calculation?
no.
axel.
Thanks answering my questions
The attachments are restart file. I am still interested in how to find problems from them.
Fan Li
coro-oh.restart.1396000 (36.5 KB)
coro-oh.restart.1434000 (36.5 KB)