Dear Alex
In my input script, I want to determine the critical temperature at which the system becomes unstable. I measure the stablility of the system by determining the number of broken bonds. I wasn’t able to accomplish this using the “pair-style” command although I need its forces effect. Therefore, I included bonds in my data file and used “bond_style harmonic with K=0” and "special_bonds lj/coul 1.0 1.0 1.0 " in order to suppress its forces effects. I put the results of “fix bond/break” in a variable to count the broken bonds and make LAMMPS decide the next temperature accordingly. The result was the system started rapturing without detecting even one broken bond. is there any mistake I made in my script to have this result
DP_A_DNT_16.data (103.0 KB)
DP_A_DNT_16_1500C.in (2.1 KB)
Sorry, but for the example provided the bond lengths don’t seem to reach 2 angstroms, so why should there be any broken bonds?
BTW: it would be even simpler to use bond style zero for this purpose. That way you don’t even compute/add zero forces.
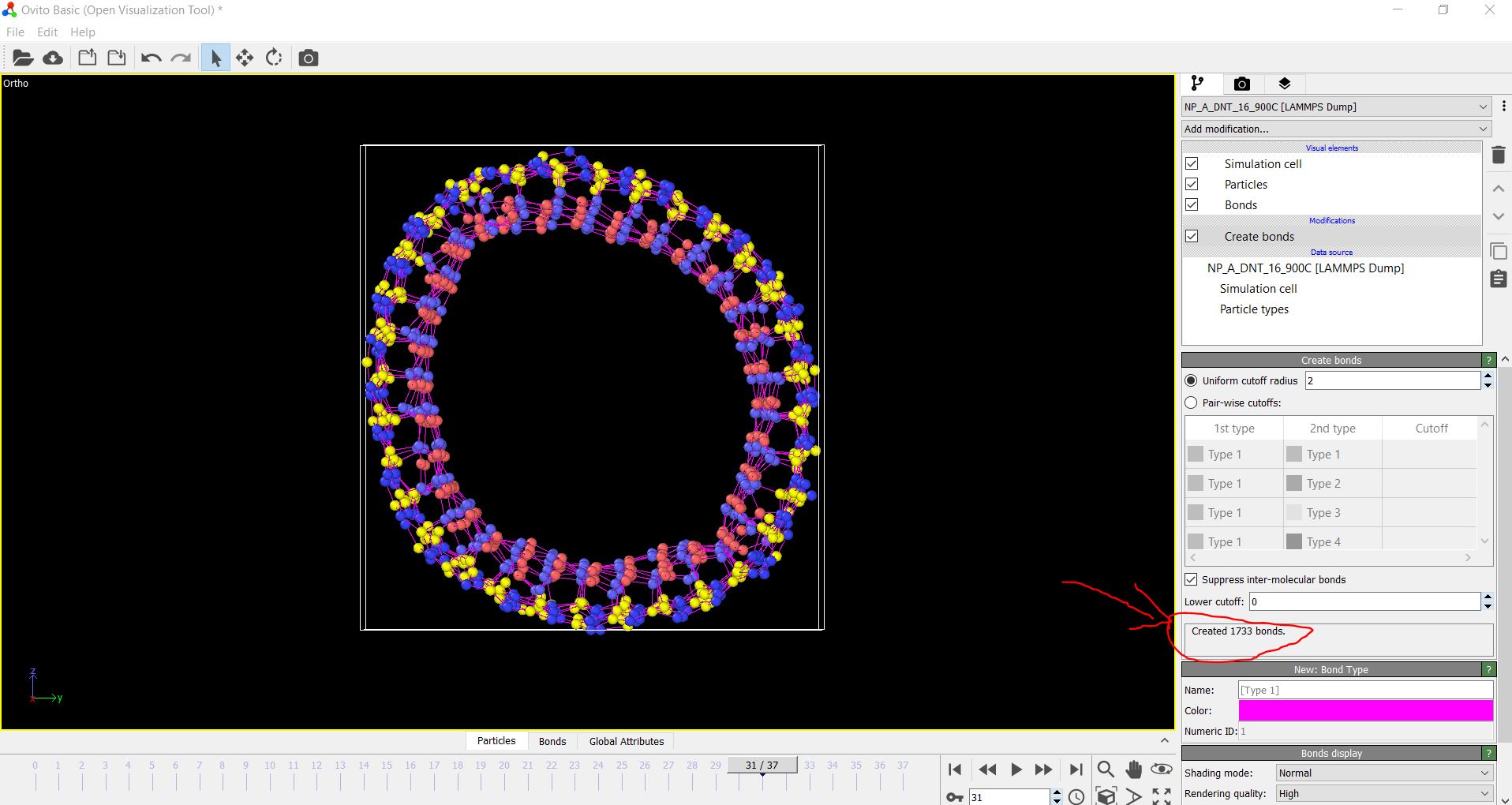
Well, that’s another weird result displayed in “thermo-style” command. I suppose it’s the longest bond for non-broken bonds. On the other hand, when I display the results of the dump file on OVITO, the number of bonds decrease rapidly indicating that there are many undetected broken bonds in LAMMPS. As shown in the attached picture, not to mention how the system raptured completely, the number of bonds reached 1733 while that number was 1840 in the data file. this means that LAMMPS was supposed to display 107 broken bonds. is it a mistake in my input file?
I also attached a more simple system with it’s results [dumped file and log file]
log.lammps (22.8 KB)
NP_A_DNT_16.data (71.1 KB)
NP_A_DNT_16_900C (2.1 MB)
NP_A_DNT_16_900C.in (2.0 KB)
Regarding the “bond-style” command, I couldn’t really understand from LAMMPS document what its coefficient stands for, is the equilibrium distance the cutoff distance for the bonds?
Please point out the confusing paragraph and the specific document.
What kind of cutoff distance are you talking about?
Please see the documentation for the variable command — LAMMPS documentation
and study carefully how you can refer to the value of other variables from inside a variable definition.
Please also see: 5.2. Parsing rules for input scripts — LAMMPS documentation
It looks to me that your computation of the total number of broken bonds is broken (pun intended). For confirmation you could just output the numbers of each fix individually.
According to the error messages, when I use the “bond_style zero” I have to specify the coefficient, what exactly does this coefficient stand for?
According to the bond_style zero command — LAMMPS documentation, it represents the equilibrium distance!!! ← this is the confusing part
You are leaving out the most important part of that explanation: , e.g. for use with fix shake.
While bond style zero does not do any computation, all bond styles need to provide an equilibrium distance so they can be constrained to that distance.
I don’t understand why that is confusing. The documentation says what it is and even if you don’t compute any forces you can assign a desired equilibrium distance. It most certainly says nothing about a cutoff or something. What I find confusing about your confusion is that you were not confused about setting the equilibrium distance for a harmonic bond with no force constant, which is effectively the same thing.
1 Like
Now it worked! As you said, the mistake was from the way I refered to the variables. Thank you very much.
1 Like