Hi,
I’m trying to simulate the T vs d curve of the Nitrogen gas using a 5 point model of the Nitrogen molecule. This model is made of 2 Nitrogen atoms, and three “ghost sites” ( one in the center and two at the ends). The N2-N2 pair potential is represented in terms of the interaction of these sites. The site-site interaction strengths are taken from this work.
I created a simple model with 1000 such N2 molecules and did rigid/NPT calculations at different temperatures to get the T vs density curve. I also simulated a 3 Point model of the Nitrogen molecule using the files given here (I extracted the tabulated form of the LJ terms given there and used the pairpotential type as tabular instead of using the inbuilt LJ function. ). I then compared the results of these two simulations to the standard data .
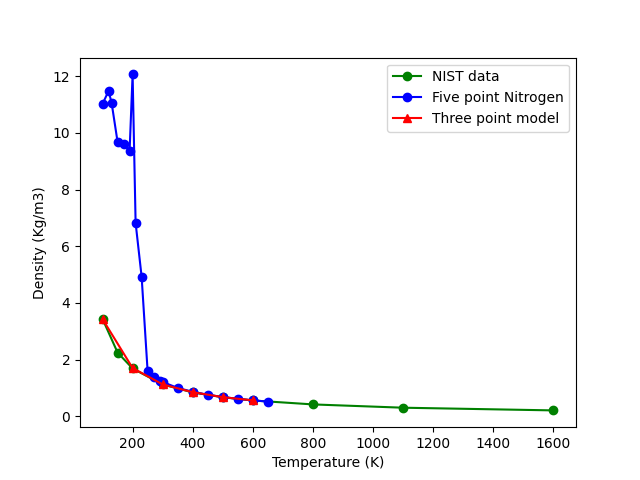 .
.
As we see here the 3 point model is giving excellent agreement to the standard values while the five point model is showing unexpected behavior below a temperature of 250 K. I’m using the same parameters and similar simulation conditions for both the 3 point and 5 point model. However, the 5 point model is giving totally wrong results, despite the fact that the 5 point model gives more accurate potential energy of the N2-N2 interaction.
Here is a comparison of the N2-N2 pair-potential of the two models. We can see that they agree quite well.
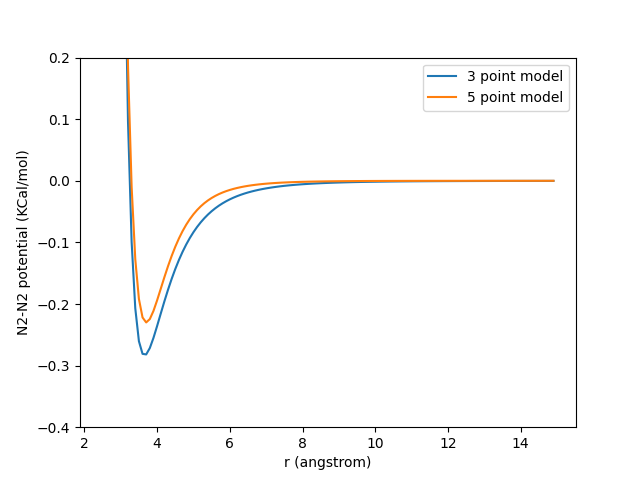 .
.
I tried so many times and can not find out what is wrong with the 5 point model, LAMMPS is not giving any errors or warnings, however the results seems to be inaccurate. For example this is the step-pressure plot of the rigid/NPT simulation of the 5 point model at T = 100K and Pressure = 0.98 atm.
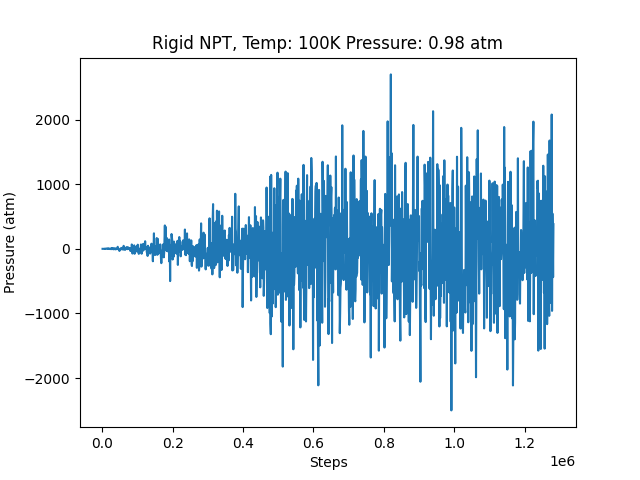 . We can see the pressure oscillating too much. also, when we observe the trajectory file, we can see that after some steps the atoms are not moving around but they are moving back and forth with respect to a point. All this suggest that there is something wrong, can anyone help me identify the issue? Here are the input files for both the 3 point and 5 point models with the respective structure files and the tabulated potential files.
. We can see the pressure oscillating too much. also, when we observe the trajectory file, we can see that after some steps the atoms are not moving around but they are moving back and forth with respect to a point. All this suggest that there is something wrong, can anyone help me identify the issue? Here are the input files for both the 3 point and 5 point models with the respective structure files and the tabulated potential files.
Is there any way to extract the molecule-molecule potential from LAMMPS? I wonder if there is any issue with my tabulated potential for the 5 point model. If I could somehow extract the potential between two rigid molecules, I could verify that the sum of the site-site interactions are leading to accurate N2-N2 potential.
Input files for the 3 point model: SimFiles3P.zip (175.7 KB)
Input files for the 5 point model:
Simfiles_5P.zip (269.5 KB)
I was working on this single issue since almost 10 days and any help and suggestions are highly appreciated!
Update: I was previously using a timestep of 1fs, I repeated the calculations, but the caklculation failed after 150,000 steps and started ouputing NaN.
LAMMPS Version: 7 Feb 2024 - Update 1
OS: Linux “Ubuntu 24.10” 6.11.0-19-generic x86_64
