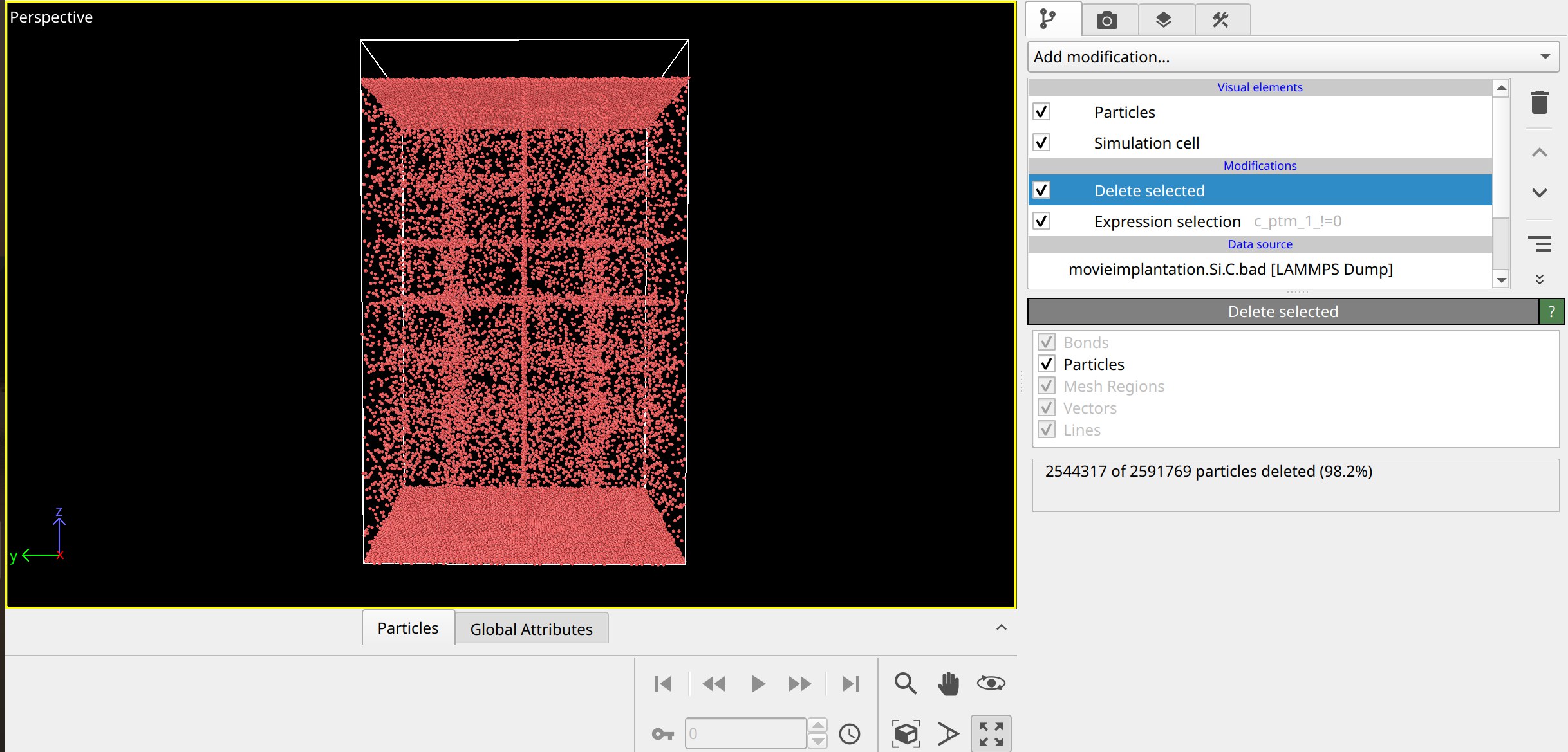
I have been trying to extract the number of defects when implanting a silicon atom into cubic diamond at 30 keV using polyhedral template matching tool in lammps. In addition, I am saving a separate file with positions of atoms that are considered defects. However, it seems that lammps incorrectly identifies the number of atoms that are not in cubic diamond structure, and editing RMSD does not seem to change anything.
Below is the script I have used, as well as the screenshot from ovito visualisation:
LAMMPS (2 Apr 2025)
boundary p p f
units metal
timestep 0.001
read_data simulations/data/relaxdata.carbon223 extra/atom/types 1
read_data simulations/data/projectiledata.Si add append shift $((xhi-xlo)/2) $((yhi-ylo)/2) $(zhi+10.0)
group projectile type 2
displace_atoms projectile random 2.5 2.5 0.0 ${seed}
pair_style hybrid/overlay tersoff zbl 6.0 7.0
pair_coeff * * tersoff simulations/potentials/C.trsf C NULL
pair_coeff 2 1 zbl 14 6
pair_coeff 2 2 zbl 14 14
#azimuthal and solid angle settings
variable energy_keV equal 30
variable projectile_mass equal 28.0855
variable energy_J equal ${energy_keV}*1.60218e-16
variable mass_kg equal ${projectile_mass}*1.6605e-27
thermo_modify lost warn flush yes
variable vel_mps equal sqrt(2*${energy_J}/${mass_kg})
variable v equal -${vel_mps}*1.0e-2
print $v
#azimuthal and solid angle settings
variable theta equal 7.0
variable vz equal ${v}*cos(${theta}*PI/180.0)
variable phi equal random(0, 360, ${seed})
variable vx atom ${v}*sin(${theta}*PI/180.0)*cos(v_phi*PI/180.0)
variable vy atom ${v}*sin(${theta}*PI/180.0)*sin(v_phi*PI/180.0)
velocity projectile set v_vx v_vy ${vz} units box
compute ek all ke/atom
compute ep all pe/atom
thermo 100
compute ptm all ptm/atom dcub 0.15 all
region interior block $(xlo+1.0) $(xhi-1.0) $(ylo+1.0) $(yhi-1.0) $(zlo+2.0) $(zhi-5.0)
region frozen block INF INF INF INF $(zlo) $(zlo+1.0)
region bottom block INF INF INF INF $(zlo+1.0) $(zlo+2.0)
region ptm_block block INF INF INF INF $(zlo+2.0) $(zhi-29.9)
region x_left block $(xlo) $(xlo+1.0) INF INF $(zlo+2.0) INF
region x_right block $(xhi-1.0) $(xhi) INF INF $(zlo+2.0) INF
region y_front block INF INF $(ylo) $(ylo+1.0) $(zlo+2.0) INF
region y_back block INF INF $(yhi-1.0) $(yhi) $(zlo+2.0) INF
region border union 5 bottom x_left x_right y_front y_back
variable non_diamond atom "c_ptm[1]==0"
group interior_atoms region interior
group frozen_atoms region frozen
group border_atoms region border
group ptm_atoms region ptm_block
group defected_atoms dynamic ptm_atoms var non_diamond
variable count_defects equal count(defected_atoms)
thermo_style custom dt time temp press v_count_defects
thermo_modify lost warn flush yes
fix 1 frozen_atoms setforce 0.0 0.0 0.0
fix 4 all dt/reset 10 NULL 0.0015 0.1 emax 2 units box
velocity frozen_atoms set 0.0 0.0 0.0
fix 2 border_atoms nvt temp 300 300 $(100.0*dt)
fix 3 interior_atoms nve
fix 5 all electron/stopping 10.0 simulations/electron_stopping/elstop.Si.C.data
variable t equal time
fix halt all halt 1 v_t >= 6.0
dump 2 defected_atoms custom 10 simulations/defects/defects.Si id x y z
dump 1 all custom 1000 simulations/movies/movieimplantation.Si.C.lammpstrj id type x y z c_ek c_ep
dump_modify 1 time yes
#run 1000000
write_data simulations/data/implantationdata.Si.C ```