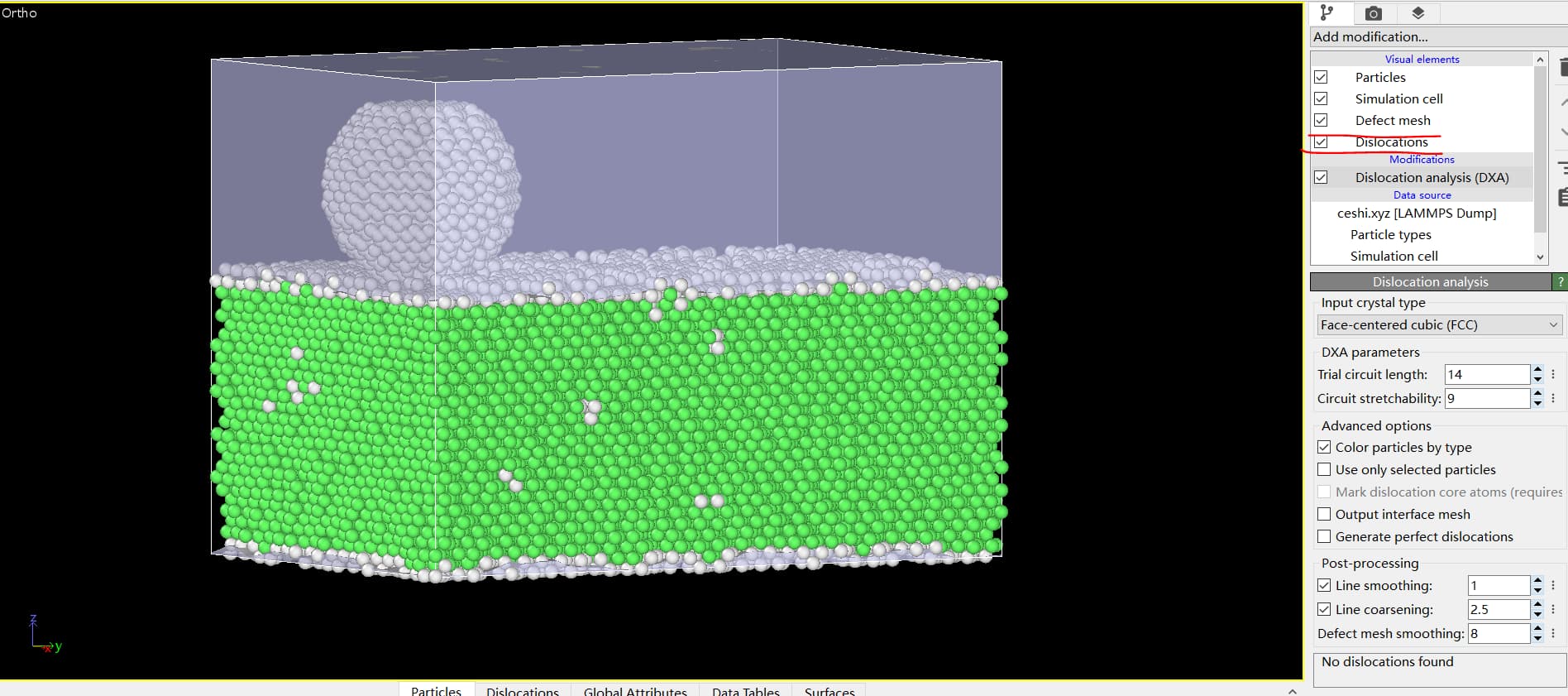
Knowledgeable experts, I encountered the following problems during the modeling process:
A smaller area was created at first, and then the change_box began to be used to expand the area along the z-axis, but some problems were found after the expansion. In the z-axis direction, there is a structure of other types instead of an FCC structure. I’d like to ask if this is an inherent feature of LAMMPS or if there’s something wrong with my modeling process
The following is my code about my model
units metal
boundary p p p
atom_style atomic
neighbor 2 bin
timestep 0.001
lattice fcc 3.56
region box block 0 20 0 30 0 13
create_box 6 box
create_atoms 1 region box
dump 77 all custom 1000 ceshi77.xyz id type x y z
set type 1 type/ratio 2 0.2 83456
set type 1 type/ratio 3 0.5 67565
set type 1 type/ratio 4 0.5 76783
set type 3 type/ratio 5 0.5 56893
mass 1 55.8
mass 2 58.7
mass 3 52.0
mass 4 59.0
mass 5 54.9
mass 6 12.0
change_box all z delta 0 35
lattice diamond 3.52
region ball sphere 10 5 18.5 4.5 #units box #x y z r
create_atoms 6 region ball
dump 99 all custom 1000 ceshi.xyz id type x y z
pair_style hybrid lj/cut 10 meam tersoff
pair_coeff * * meam library.meam Co Ni Cr Fe Mn CoNiCrFeMn.meam Co Ni Cr Fe Mn NULL
pair_coeff * * tersoff SiC.tersoff NULL NULL NULL NULL NULL C
pair_coeff 1 6 lj/cut 0.038687 2.8605
pair_coeff 2 6 lj/cut 0.038429 2.841
pair_coeff 3 6 lj/cut 0.037758 2.868
pair_coeff 4 6 lj/cut 0.038281 2.842
pair_coeff 5 6 lj/cut 0.003457 3.1325
#pair_coeff 6 6 none
dump 66 all custom 1000 ceshistr.xyz id type x y z
thermo 1000
thermo_style custom step temp pe lx ly lz
dump 1 all atom 50 mini.xyz
min_style cg
minimize 1e-10 1e-10 5000 5000
undump 1
reset_timestep 0