Dear All,
I have a question about my system, which includes protein molecules, water, and a carbon nanotube (CNT). When I run the script with a small number of molecules as a test, the molecular structures all seem to look fine:
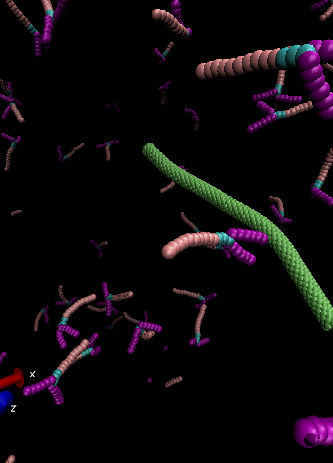
However, when I include the correct number of water molecules (~3,000,000), it seems that their structure deforms in an unexpected or incorrect way. Since generating that many water beads takes a long time, I created them in a separate LAMMPS run and saved them as a data file. In my current input script, I read in that data file and then use the delete_atoms command to remove any overlapping water molecules.

I thought it might be helpful to share the following information—I’m getting some image flag warnings along with a few others. In case these messages provide any clues, I wanted to include them here for your insight.
LAMMPS (2 Aug 2023)
OMP_NUM_THREADS environment is not set. Defaulting to 1 thread. (src/comm.cpp:98)
using 1 OpenMP thread(s) per MPI task
Reading data file …
orthogonal box = (-50 -50 -50) to (50 50 70)
2 by 2 by 5 MPI processor grid
reading atoms …
640 atoms
reading velocities …
640 velocities
scanning bonds …
5 = max bonds/atom
scanning angles …
10 = max angles/atom
reading bonds …
2224 bonds
reading angles …
6288 angles
Finding 1-2 1-3 1-4 neighbors …
special bond factors lj: 0 0 0
special bond factors coul: 0 0 0
7 = max # of 1-2 neighbors
42 = max # of 1-3 neighbors
294 = max # of 1-4 neighbors
47 = max # of special neighbors
special bonds CPU = 0.007 seconds
read_data CPU = 0.041 seconds
Reading data file …
orthogonal box = (-50 -50 -50) to (50 50 70)
2 by 2 by 5 MPI processor grid
reading atoms …
1250 atoms
scanning bonds …
2 = max bonds/atom
scanning angles …
3 = max angles/atom
reading bonds …
1200 bonds
reading angles …
1200 angles
Finding 1-2 1-3 1-4 neighbors …
special bond factors lj: 0 0 0
special bond factors coul: 0 0 0
7 = max # of 1-2 neighbors
42 = max # of 1-3 neighbors
294 = max # of 1-4 neighbors
47 = max # of special neighbors
special bonds CPU = 0.006 seconds
read_data CPU = 0.015 seconds
Reading data file …
orthogonal box = (-50 -50 -50) to (50 50 70)
2 by 2 by 5 MPI processor grid
reading atoms …
3598110 atoms
reading velocities …
3598110 velocities
Finding 1-2 1-3 1-4 neighbors …
special bond factors lj: 0 0 0
special bond factors coul: 0 0 0
7 = max # of 1-2 neighbors
42 = max # of 1-3 neighbors
294 = max # of 1-4 neighbors
47 = max # of special neighbors
special bonds CPU = 0.104 seconds
read_data CPU = 25.464 seconds
3598110 atoms in group liquid
640 atoms in group CNT
500 atoms in group PEG
600 atoms in group DSPEt
150 atoms in group DSPEH
1140 atoms in group CNTPEG
750 atoms in group DSPE
1890 atoms in group DCP
1250 atoms in group DSPEG
System init for delete_atoms …
Generated 0 of 10 mixed pair_coeff terms from geometric mixing rule
Neighbor list info …
update: every = 1 steps, delay = 0 steps, check = yes
max neighbors/atom: 2000, page size: 100000
master list distance cutoff = 3.58
ghost atom cutoff = 6
binsize = 1.79, bins = 56 56 68
2 neighbor lists, perpetual/occasional/extra = 1 1 0
(1) command delete_atoms, occasional
attributes: full, newton on
pair build: full/bin
stencil: full/bin/3d
bin: standard
(2) pair edpd, perpetual
attributes: half, newton on
pair build: half/bin/newton
stencil: half/bin/3d
bin: standard
WARNING: Ignoring ‘compress yes’ for molecular system (src/delete_atoms.cpp:140)
Deleted 1915 atoms, new total = 3598085
System init for delete_atoms …
Generated 0 of 10 mixed pair_coeff terms from geometric mixing rule
Neighbor list info …
update: every = 1 steps, delay = 0 steps, check = yes
max neighbors/atom: 2000, page size: 100000
master list distance cutoff = 3.58
ghost atom cutoff = 6
binsize = 1.79, bins = 56 56 68
2 neighbor lists, perpetual/occasional/extra = 1 1 0
(1) command delete_atoms, occasional
attributes: full, newton on
pair build: full/bin
stencil: full/bin/3d
bin: standard
(2) pair edpd, perpetual
attributes: half, newton on
pair build: half/bin/newton
stencil: half/bin/3d
bin: standard
WARNING: Ignoring ‘compress yes’ for molecular system (src/delete_atoms.cpp:140)
Deleted 6599 atoms, new total = 3591486
System init for delete_atoms …
Generated 0 of 10 mixed pair_coeff terms from geometric mixing rule
Neighbor list info …
update: every = 1 steps, delay = 0 steps, check = yes
max neighbors/atom: 2000, page size: 100000
master list distance cutoff = 3.58
ghost atom cutoff = 6
binsize = 1.79, bins = 56 56 68
2 neighbor lists, perpetual/occasional/extra = 1 1 0
(1) command delete_atoms, occasional
attributes: full, newton on
pair build: full/bin
stencil: full/bin/3d
bin: standard
(2) pair edpd, perpetual
attributes: half, newton on
pair build: half/bin/newton
stencil: half/bin/3d
bin: standard
WARNING: Ignoring ‘compress yes’ for molecular system (src/delete_atoms.cpp:140)
Deleted 4308 atoms, new total = 3587178
3586538 atoms in group nonrigid
1 rigid bodies with 640 atoms
CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE
Your simulation uses code contributions which should be cited:
- pair edpd command: doi:10.1016/j.jcp.2014.02.003
The log file lists these citations in BibTeX format.
CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE
Generated 0 of 10 mixed pair_coeff terms from geometric mixing rule
Neighbor list info …
update: every = 1 steps, delay = 0 steps, check = yes
max neighbors/atom: 2000, page size: 100000
master list distance cutoff = 2.58
ghost atom cutoff = 6
binsize = 1.29, bins = 78 78 94
1 neighbor lists, perpetual/occasional/extra = 1 0 0
(1) pair edpd, perpetual
attributes: half, newton on
pair build: half/bin/newton
stencil: half/bin/3d
bin: standard
Setting up Verlet run …
Unit style : lj
Current step : 0
Time step : 0.005
WARNING: Inconsistent image flags (src/domain.cpp:815)
Per MPI rank memory allocation (min/avg/max) = 497.3 | 498 | 498.9 Mbytes
Step Temp E_pair E_mol TotEng Press
0 1.0004646 42.063095 0.0068759588 43.5704 146.98242
1000 1.0038585 41.898826 0.013351094 43.417696 146
I’d really appreciate it if you could share your thoughts based on your experience. Do you think the issue might just be due to incorrect image flags? If so, could you please advise me on how to fix them? Or do you suspect there might be other potential causes I should look into?
my simulation got killed :
78
3000 1.0031866 41.896949 0.013396446 43.414857 146.36105
4000 1.0031595 41.896872 0.013524909 43.414868 146.16797
ERROR on proc 3: Bond atoms 1595 1596 missing on proc 3 at step 4190 (src/ntopo_bond_all.cpp:59)
Last command: run 10000000
MPI_ABORT was invoked on rank 3 in communicator MPI_COMM_WORLD
Proc: [[65140,1],3]
Errorcode: 1
NOTE: invoking MPI_ABORT causes Open MPI to kill all MPI processes.
You may or may not see output from other processes, depending on
exactly when Open MPI kills them.
Here is the logfile:
" LAMMPS (2 Aug 2023)
OMP_NUM_THREADS environment is not set. Defaulting to 1 thread. (src/comm.cpp:98)
using 1 OpenMP thread(s) per MPI task
units lj
dimension 3
timestep 0.005
boundary p p p
neighbor 2 bin
neigh_modify every 1 delay 0 check yes
comm_modify vel yes
comm_modify cutoff 6.0
atom_style hybrid edpd full
bond_style harmonic
angle_style harmonic
pair_style edpd 1.58 9872598
region mybox block -50 50 -50 50 -50 70 units box
read_data equi.data
Reading data file ...
orthogonal box = (-50 -50 -50) to (50 50 70)
2 by 2 by 5 MPI processor grid
reading atoms ...
640 atoms
reading velocities ...
640 velocities
scanning bonds ...
5 = max bonds/atom
scanning angles ...
10 = max angles/atom
reading bonds ...
2224 bonds
reading angles ...
6288 angles
Finding 1-2 1-3 1-4 neighbors ...
special bond factors lj: 0 0 0
special bond factors coul: 0 0 0
7 = max # of 1-2 neighbors
42 = max # of 1-3 neighbors
294 = max # of 1-4 neighbors
47 = max # of special neighbors
special bonds CPU = 0.008 seconds
read_data CPU = 0.044 seconds
read_data DSPEpeg_full1.data add append
Reading data file ...
orthogonal box = (-50 -50 -50) to (50 50 70)
2 by 2 by 5 MPI processor grid
reading atoms ...
1250 atoms
scanning bonds ...
2 = max bonds/atom
scanning angles ...
3 = max angles/atom
reading bonds ...
1200 bonds
reading angles ...
1200 angles
Finding 1-2 1-3 1-4 neighbors ...
special bond factors lj: 0 0 0
special bond factors coul: 0 0 0
7 = max # of 1-2 neighbors
42 = max # of 1-3 neighbors
294 = max # of 1-4 neighbors
47 = max # of special neighbors
special bonds CPU = 0.006 seconds
read_data CPU = 0.015 seconds
read_data waterbeads.data add append
Reading data file ...
orthogonal box = (-50 -50 -50) to (50 50 70)
2 by 2 by 5 MPI processor grid
reading atoms ...
3598110 atoms
reading velocities ...
3598110 velocities
Finding 1-2 1-3 1-4 neighbors ...
special bond factors lj: 0 0 0
special bond factors coul: 0 0 0
7 = max # of 1-2 neighbors
42 = max # of 1-3 neighbors
294 = max # of 1-4 neighbors
47 = max # of special neighbors
special bonds CPU = 0.111 seconds
read_data CPU = 25.633 seconds
#fix recenter all recenter INIT INIT INIT units box
# create water beads (type 2)
#print ">>> Creating water"
#create_atoms 2 random 3598110 13487 NULL overlap 0.4 maxtry 500
#create_atoms 2 random 35 13487 NULL overlap 0.4 maxtry 500
#print ">>> Water created"
# group the newly created water beads
group liquid type 2
3598110 atoms in group liquid
# GROUPING
group CNT type 1
640 atoms in group CNT
group PEG type 3
500 atoms in group PEG
group DSPEt type 4
600 atoms in group DSPEt
group DSPEH type 5
150 atoms in group DSPEH
group CNTPEG union CNT PEG
1140 atoms in group CNTPEG
group DSPE union DSPEt DSPEH
750 atoms in group DSPE
group DCP union DSPE CNTPEG
1890 atoms in group DCP
group DSPEG union DSPE PEG
1250 atoms in group DSPEG
# pair, bond & angle coeff
include parmcnt.lammps
pair_coeff 1 1 value 4.5 0.41 1.58 1.41E-5 2.0 1.58 #(C-C)
pair_coeff 2 2 value 4.5 0.41 1.58 1.41E-5 2.0 1.58 # (W-W)
pair_coeff 1 2 value 4.5 0.41 1.58 1.41E-5 2.0 1.58 # (C-W)
bond_coeff 1 value value
bond_coeff 2 value value
angle_coeff 1 value value
angle_coeff 2 value value
angle_coeff 3 value value
mass 1 value #5 Carbon atoms per bead
mass 2 value #3 water molecule per bead
include parmPEG.lammps
pair_coeff 3 3 value 4.5 0.41 1.58 1.41E-5 2.0 1.58 #PEG-PEG
pair_coeff 3 1 value 4.5 0.41 1.58 1.41E-5 2.0 1.58 #PEG-CARBON
pair_coeff 3 2 value 4.5 0.41 1.58 1.41E-5 2.0 1.58 #PEG-Water.
bond_coeff 3 150 0.75
angle_coeff 4 30 180
mass 3 0.815 # 1 PEG monomer per bead (reduced mass of PEG= mass of PEG monomer/ mass of 3 water molecules)
include parmDSPE.lammps
pair_coeff 4 4 value 4.5 0.41 1.58 1.41E-5 2.0 1.58 #DSPEt-DSPEt
pair_coeff 5 5 value 4.5 0.41 1.58 1.41E-5 2.0 1.58 #DSPEH-DSPEH
pair_coeff 4 5 value 4.5 0.41 1.58 1.41E-5 2.0 1.58 #DSPEH-DSPEt
pair_coeff 4 1 value 4.5 0.41 1.58 1.41E-5 2.0 1.58 #DSPEt-CARBON
pair_coeff 5 1 value 4.5 0.41 1.58 1.41E-5 2.0 1.58 #DSPEH-CARBON
pair_coeff 4 2 value 4.5 0.41 1.58 1.41E-5 2.0 1.58 #DSPEt-Water
pair_coeff 5 2 value 4.5 0.41 1.58 1.41E-5 2.0 1.58 #DSPEH-Water
pair_coeff 4 3 value 4.5 0.41 1.58 1.41E-5 2.0 1.58 #DSPEt-PEG
pair_coeff 5 3 value 4.5 0.41 1.58 1.41E-5 2.0 1.58 #DSPEH-PEG
bond_coeff 4 value value
bond_coeff 5 value value
bond_coeff 6 value value
angle_coeff 5 value value
angle_coeff 6 value value
angle_coeff 7 value value #used to be 150
angle_coeff 8 value value
angle_coeff 9 value value
angle_coeff 10 value value
mass 4 0.815
mass 5 0.815
# remove water beads that overlap with CNT, DSPE, or PEG
delete_atoms overlap 1.0 liquid CNT
System init for delete_atoms ...
Generated 0 of 10 mixed pair_coeff terms from geometric mixing rule
Neighbor list info ...
update: every = 1 steps, delay = 0 steps, check = yes
max neighbors/atom: 2000, page size: 100000
master list distance cutoff = 3.58
ghost atom cutoff = 6
binsize = 1.79, bins = 56 56 68
2 neighbor lists, perpetual/occasional/extra = 1 1 0
(1) command delete_atoms, occasional
attributes: full, newton on
pair build: full/bin
stencil: full/bin/3d
bin: standard
(2) pair edpd, perpetual
attributes: half, newton on
pair build: half/bin/newton
stencil: half/bin/3d
bin: standard
WARNING: Ignoring 'compress yes' for molecular system (src/delete_atoms.cpp:140)
Deleted 1915 atoms, new total = 3598085
delete_atoms overlap 1.0 liquid DSPE
System init for delete_atoms ...
Generated 0 of 10 mixed pair_coeff terms from geometric mixing rule
Neighbor list info ...
update: every = 1 steps, delay = 0 steps, check = yes
max neighbors/atom: 2000, page size: 100000
master list distance cutoff = 3.58
ghost atom cutoff = 6
binsize = 1.79, bins = 56 56 68
2 neighbor lists, perpetual/occasional/extra = 1 1 0
(1) command delete_atoms, occasional
attributes: full, newton on
pair build: full/bin
stencil: full/bin/3d
bin: standard
(2) pair edpd, perpetual
attributes: half, newton on
pair build: half/bin/newton
stencil: half/bin/3d
bin: standard
WARNING: Ignoring 'compress yes' for molecular system (src/delete_atoms.cpp:140)
Deleted 6561 atoms, new total = 3591524
delete_atoms overlap 1.0 liquid PEG
System init for delete_atoms ...
Generated 0 of 10 mixed pair_coeff terms from geometric mixing rule
Neighbor list info ...
update: every = 1 steps, delay = 0 steps, check = yes
max neighbors/atom: 2000, page size: 100000
master list distance cutoff = 3.58
ghost atom cutoff = 6
binsize = 1.79, bins = 56 56 68
2 neighbor lists, perpetual/occasional/extra = 1 1 0
(1) command delete_atoms, occasional
attributes: full, newton on
pair build: full/bin
stencil: full/bin/3d
bin: standard
(2) pair edpd, perpetual
attributes: half, newton on
pair build: half/bin/newton
stencil: half/bin/3d
bin: standard
WARNING: Ignoring 'compress yes' for molecular system (src/delete_atoms.cpp:140)
Deleted 4146 atoms, new total = 3587378
# THERMOSTATS FOR CNT AND LIQUID GROUPS
compute tliquid liquid temp
compute tCNT CNT temp
compute tDSPE DSPE temp
compute tPEG PEG temp
compute tDSPEG DSPEG temp
group nonrigid subtract all CNT
3586738 atoms in group nonrigid
fix mynve nonrigid nve
fix rigidCNT CNT rigid single
1 rigid bodies with 640 atoms
#fix mynve all nve
#fix rigidCNT CNT rigid single
neighbor 1 bin
neigh_modify delay 0 every 1 check yes
comm_modify mode single cutoff 6.0 vel yes
fix mylang1 liquid langevin 1 1 10 8786
fix_modify mylang1 temp tliquid
#fix mylang2 CNT langevin 2 2 10 7768
#fix_modify mylang2 temp tCNT
fix mylang3 DSPEG langevin 1 1 10 7730
fix_modify mylang3 temp tDSPEG
fix tetherCNT CNT spring/self 50.0
velocity CNT set 0.0 0.0 0.0
# EQUILIBRATE WITH THERMOSTATS
run 1000
CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE
Your simulation uses code contributions which should be cited:
- pair edpd command: doi:10.1016/j.jcp.2014.02.003
@Article{ZLi2014_JCP,
author = {Li, Z. and Tang, Y.-H. and Lei, H. and Caswell, B. and Karniadakis, G. E.},
title = {Energy-Conserving Dissipative Particle Dynamics with Temperature-Dependent Properties},
journal = {Journal of Computational Physics},
year = {2014},
volume = {265},
pages = {113--127}
}
@Article{ZLi2015_CC,
author = {Li, Z. and Tang, Y.-H. and Li, X. and Karniadakis, G. E.},
title = {Mesoscale Modeling of Phase Transition Dynamics of Thermoresponsive Polymers},
journal = {Chemical Communications},
year = {2015},
volume = {51},
pages = {11038--11040}
}
CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE
Generated 0 of 10 mixed pair_coeff terms from geometric mixing rule
Neighbor list info ...
update: every = 1 steps, delay = 0 steps, check = yes
max neighbors/atom: 2000, page size: 100000
master list distance cutoff = 2.58
ghost atom cutoff = 6
binsize = 1.29, bins = 78 78 94
1 neighbor lists, perpetual/occasional/extra = 1 0 0
(1) pair edpd, perpetual
attributes: half, newton on
pair build: half/bin/newton
stencil: half/bin/3d
bin: standard
WARNING: Inconsistent image flags (src/domain.cpp:815)
Per MPI rank memory allocation (min/avg/max) = 497 | 498 | 498.9 Mbytes
Step Temp E_pair E_mol TotEng Press
0 1.0004067 42.063971 0.0068755708 43.571189 146.99341
1000 1.0024538 41.901957 0.014429364 43.419799 146.33634
Loop time of 1097.78 on 20 procs for 1000 steps with 3587378 atoms... "
`````````````````````