Hi LAMMPS users,
I noticed a change in behavior between a data set run with the stable 29 Sep 2021 build of LAMMPS and the stable 23 Jun 2022 build, with supporting images in the pdf.
LAMMPS 2021 2022 Stress Strain Behavior Change.pdf (318.4 KB)
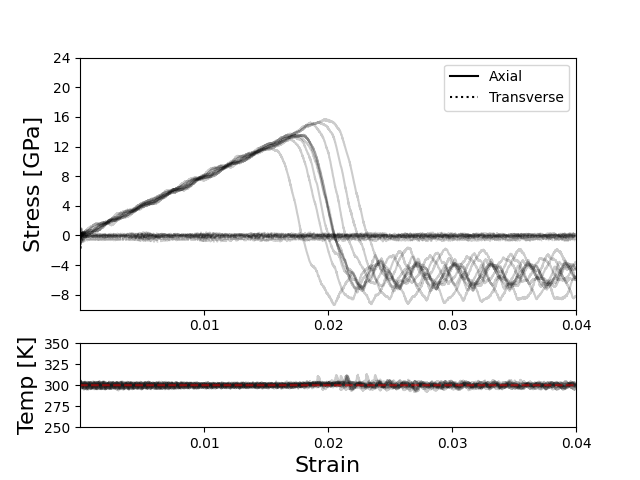
The set up is a graphene sheet with a hole in it, shown in Figure 1, sampled at 100 points in time after a 5 ns equilibration. Figure 2 contains the first ten restarts for each set of damping parameter combinations, with Figures 2B and 2C being the same input (below) on the same data using the June 2022 and Sep 2021 LAMMPS builds, respectively. Figure 2A is an NVT run and Figure 2D an NPT with the standard ‘rule of thumb’ damping parameter values for reference. The behavior of the Jun 2022 build is consistent under slightly different (but effectively the same) damping parameters, with the transverse stress stable at some value around 0 GPa (see extra image below), compared to the relatively volatile transverse values returned by the Sep 2021 build.
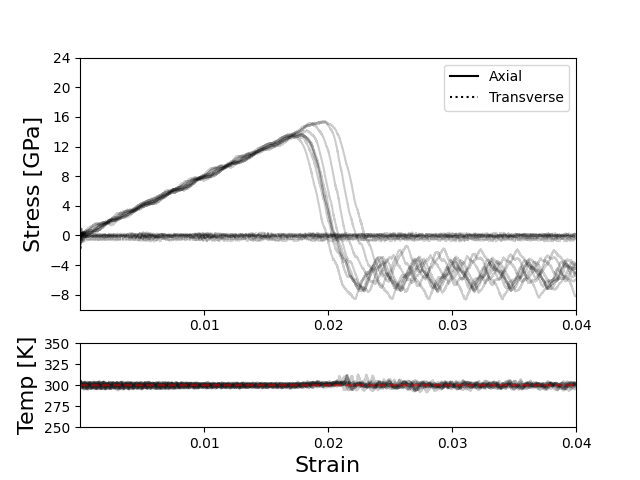
Figure 3 plots both behaviors as failure probabilities, with the transverse behaviors in Figure 3B sampled at the ultimate axial stress value used in Figure 3A. These distributions use all 100 restarts generated from the graphene sheet.
I’ve looked through the change logs and related src files on github to see if there were any changes that might lead to this difference in behavior, but I could not find anything that stood out. All restarts, starting data, and the REBO potential used were either developed using, or from, the 29 Sept 2021 build, so it could be a backwards compatibility issue on my part that I missed when looking at the documentation.
Does anyone have any ideas as to what might be causing this?
Thanks,
Tj
Edit: Added a restart file and REBO potential
LuoArmchairSingleCrystal-a0-10-r02_1.rst (8.5 MB)
CH_Luo.rebo (905.0 KB)
Tensile code for reference, with command line input:
# Command line input values
mpirun -np 6 --bind-to socket -x OMP_NUM_THREADS=2 lmp -sf hybrid gpu omp -pk gpu 1 gpuID 0 -in 20220517_Graphene-Tensile.in
#-----------------------------------------------------------
# Simualtion Variables
#-----------------------------------------------------------
# Temperature
variable T1 equal 300
# Timestep
variable tms equal 0.001 # ps
variable tdamp equal ${tms}*100.0
variable pdamp equal ${tms}*999.0
#-----------------------------------------------------------------
# Graphene tensile
#-----------------------------------------------------------------
units metal
atom_style molecular
variable fname string 'LuoArmchairSingleCrystal-a0-10-r02'
label equi_timesteps
variable equi_loop loop 1 100
variable loop_val equal ${equi_loop}
read_restart ${fname}_${loop_val}.rst
pair_style rebo #1.92
pair_coeff * * CH_Luo.rebo C
mass * 12.0107 #g/mol
newton on
#-----------------------------------------------------------
# Set up computes
#-----------------------------------------------------------
compute temps all temp
# Stress/ Atom Method
compute SAperatom all stress/atom NULL #virial
# average per atom. (Summing and dividing over total volume) and (averaging peratom stress then dividing by single volume) are the same
compute SApress all reduce ave c_SAperatom[1] c_SAperatom[2] c_SAperatom[3] c_SAperatom[4] c_SAperatom[5] c_SAperatom[6]
#-----------------------------------------------------------
# Run Equilibration
#-----------------------------------------------------------
# fix 1 all npt temp ${T1} ${T1} ${tdamp} x 1.01325 1.01325 ${pdamp} z 0.0 0.0 ${pdamp}
#
# thermo_style custom step temp
# thermo 1000
# timestep ${tms}
# run 100000
#-----------------------------------------------------------
# Run Tensile
#-----------------------------------------------------------
# Set up
variable temps equal c_temps
variable tmpx equal "lx"
variable tmpy equal "ly"
variable Lx0 equal ${tmpx}
variable Ly0 equal ${tmpy}
# Strain
variable strainx equal "(lx - v_Lx0)/v_Lx0"
variable strainy equal "(ly - v_Ly0)/v_Ly0"
variable truestrainx equal "ln(lx/v_Lx0)"
variable truestrainy equal "ln(ly/v_Ly0)"
# graphene volume
variable xyarea equal "lx*ly"
variable graph_thickness equal 3.4 # taken from Luo Paper
variable totalbeads equal count(all)
variable graph_vol equal "v_xyarea*v_graph_thickness/v_totalbeads"
# area ratio
variable aratio equal "v_xyarea/(v_Lx0*v_Ly0)"
# sampling rate
variable sample_rate equal 10
# stress/atom is units pressure*volume, so divide by volume, and convert bar to Pa
variable SA_stressxx equal c_SApress[1]/v_graph_vol*1E5
variable SA_stressyy equal c_SApress[2]/v_graph_vol*1E5
variable SA_stresszz equal c_SApress[3]/v_graph_vol*1E5
variable SA_stressxy equal c_SApress[4]/v_graph_vol*1E5
variable SA_stressxz equal c_SApress[5]/v_graph_vol*1E5
variable SA_stressyz equal c_SApress[6]/v_graph_vol*1E5
# out
fix def21 all print ${sample_rate} "${strainy} ${truestrainy} ${SA_stressxx} ${SA_stressyy} ${SA_stresszz} ${SA_stressxy} ${SA_stressxz} ${SA_stressyz}" file Graphene_NPT_REBO_Strain.SA${sample_rate}_${loop_val}.txt screen no
fix def22 all print ${sample_rate} "${temps} ${strainy} ${aratio} ${SA_stressxx} ${SA_stressyy} ${SA_stresszz}" file Graphene_NPT_TEMP_REBO_Strain.SA${sample_rate}_${loop_val}.txt screen no
dump 14 all custom 100 Graphene_NPT_REBO_Strain.dump id mol type x y z c_SAperatom[*]
# fixes and run
# fix 1 all nvt temp ${T1} ${T1} ${tdamp}
fix 1 all npt temp ${T1} ${T1} ${tdamp} x 1.01325 1.01325 ${pdamp} z 0.0 0.0 ${pdamp}
fix 2 all deform 1 y erate ${tms}
thermo_style custom step temp v_strainy v_SA_stressyy v_aratio
thermo 100
timestep ${tms}
run 40000
clear
next equi_loop
jump SELF equi_timesteps