Dear Sir,
My file upon compilation in LAMMPS shows the following error :-
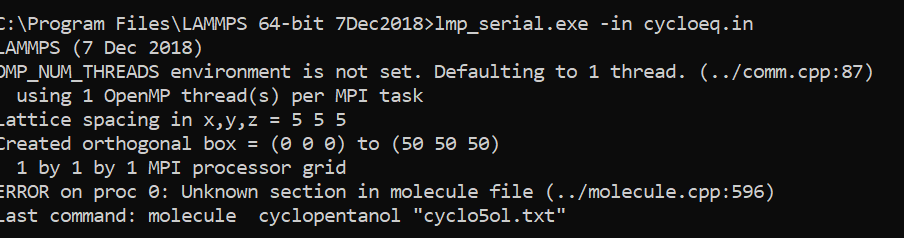
Could you please help me with this?
‘Unknown section in molecule file’.
Here is the molecule file…
16 atoms
16 bonds
31 angles
48 dihedrals
10 impropers
16 atom types
16 bond types
31 angle types
48 dihedral types
10 improper types
-1.874300 48.125700 xlo xhi
-0.959350 49.040650 ylo yhi
-0.959590 49.040410 zlo zhi
Masses
1 12.011
2 12.011
3 12.011
4 12.011
5 12.011
6 15.999
7 1.008
8 1.008
9 1.008
10 1.008
11 1.008
12 1.008
13 1.008
14 1.008
15 1.008
16 1.008
Pair Coeffs
1 0.066 3.5000000
2 0.066 3.5000000
3 0.066 3.5000000
4 0.066 3.5000000
5 0.066 3.5000000
6 0.170 3.1200000
7 0.030 2.5000000
8 0.030 2.5000000
9 0.030 2.5000000
10 0.030 2.5000000
11 0.030 2.5000000
12 0.030 2.5000000
13 0.030 2.5000000
14 0.030 2.5000000
15 0.030 2.5000000
16 0.000 0.0000000
Bond Coeffs
1 268.0000 1.5290
2 268.0000 1.5290
3 268.0000 1.5290
4 268.0000 1.5290
5 320.0000 1.4100
6 340.0000 1.0900
7 340.0000 1.0900
8 340.0000 1.0900
9 340.0000 1.0900
10 340.0000 1.0900
11 340.0000 1.0900
12 340.0000 1.0900
13 340.0000 1.0900
14 340.0000 1.0900
15 553.0000 0.9450
16 268.0000 1.5290
Angle Coeffs
1 58.350 112.700
2 58.350 112.700
3 58.350 112.700
4 50.000 109.500
5 37.500 110.700
6 37.500 110.700
7 37.500 110.700
8 37.500 110.700
9 37.500 110.700
10 37.500 110.700
11 37.500 110.700
12 37.500 110.700
13 37.500 110.700
14 55.000 108.500
15 50.000 109.500
16 37.500 110.700
17 37.500 110.700
18 33.000 107.800
19 33.000 107.800
20 37.500 110.700
21 37.500 110.700
22 58.350 112.700
23 33.000 107.800
24 37.500 110.700
25 37.500 110.700
26 35.000 109.500
27 37.500 110.700
28 37.500 110.700
29 33.000 107.800
30 58.350 112.700
31 37.500 110.700
Dihedral Coeffs
1 1.300 -0.200 0.200 0.000
2 1.300 -0.200 0.200 0.000
3 -0.356 -0.174 0.492 0.000
4 1.300 -0.200 0.200 0.000
5 0.000 0.000 0.300 0.000
6 0.000 0.000 0.300 0.000
7 -1.552 0.000 0.000 0.000
8 0.000 0.000 0.300 0.000
9 0.000 0.000 0.300 0.000
10 0.000 0.000 0.300 0.000
11 0.000 0.000 0.300 0.000
12 0.000 0.000 0.300 0.000
13 0.000 0.000 0.300 0.000
14 0.000 0.000 0.300 0.000
15 0.000 0.000 0.352 0.000
16 0.000 0.000 0.300 0.000
17 -1.552 0.000 0.000 0.000
18 0.000 0.000 0.300 0.000
19 -0.356 -0.174 0.492 0.000
20 0.000 0.000 0.300 0.000
21 0.000 0.000 0.300 0.000
22 0.000 0.000 0.300 0.000
23 0.000 0.000 0.300 0.000
24 0.000 0.000 0.300 0.000
25 0.000 0.000 0.300 0.000
26 0.000 0.000 0.300 0.000
27 0.000 0.000 0.300 0.000
28 0.000 0.000 0.468 0.000
29 0.000 0.000 0.300 0.000
30 0.000 0.000 0.300 0.000
31 0.000 0.000 0.468 0.000
32 1.300 -0.200 0.200 0.000
33 0.000 0.000 0.300 0.000
34 0.000 0.000 0.300 0.000
35 1.300 -0.200 0.200 0.000
36 0.000 0.000 0.300 0.000
37 0.000 0.000 0.300 0.000
38 0.000 0.000 0.300 0.000
39 0.000 0.000 0.300 0.000
40 0.000 0.000 0.300 0.000
41 0.000 0.000 0.300 0.000
42 0.000 0.000 0.300 0.000
43 0.000 0.000 0.300 0.000
44 0.000 0.000 0.468 0.000
45 0.000 0.000 0.468 0.000
46 0.000 0.000 0.300 0.000
47 0.000 0.000 0.300 0.000
48 0.000 0.000 0.300 0.000
Improper Coeffs
1 0.000 -1 2
2 0.000 -1 2
3 0.000 -1 2
4 0.000 -1 2
5 0.000 -1 2
6 0.000 -1 2
7 0.000 -1 2
8 0.000 -1 2
9 0.000 -1 2
10 0.000 -1 2
Atoms
1 1 1 -0.18760000 1.000 1.00000 0.00000
2 1 2 -0.18470000 -0.520 1.00000 0.00000
3 1 3 -0.22290000 -0.854 1.00000 1.48150
4 1 4 0.06970000 0.187 0.06764 2.09766
5 1 5 -0.22880000 1.374 0.05277 1.13652
6 1 6 -0.58160000 0.571 0.52195 3.38495
7 1 7 0.09400000 1.374 2.00753 0.21804
8 1 8 0.09400000 1.417 0.68136 -0.95959
9 1 9 0.09760000 -0.900 0.09000 -0.47832
10 1 10 0.09760000 -0.936 1.86690 -0.52121
11 1 11 0.10210000 -1.874 0.65069 1.66998
12 1 12 0.10210000 -0.773 2.01259 1.89493
13 1 13 0.13280000 -0.218 -0.94432 2.20535
14 1 14 0.10450000 1.499 -0.95935 0.73371
15 1 15 0.10450000 2.318 0.34418 1.60840
16 1 16 0.40690000 0.966 1.40503 3.28568
Bonds
1 1 2 1
2 2 3 2
3 3 4 3
4 4 5 1
5 5 6 4
6 6 7 1
7 7 8 1
8 8 9 2
9 9 10 2
10 10 11 3
11 11 12 3
12 12 13 4
13 13 14 5
14 14 15 5
15 15 16 6
16 16 5 4
Angles
1 1 1 2 3
2 2 2 3 4
3 3 2 1 5
4 4 3 4 6
5 5 2 1 7
6 6 2 1 8
7 7 1 2 9
8 8 1 2 10
9 9 2 3 11
10 10 2 3 12
11 11 3 4 13
12 12 1 5 14
13 13 1 5 15
14 14 4 6 16
15 15 5 4 6
16 16 4 3 11
17 17 5 1 8
18 18 9 2 10
19 19 11 3 12
20 20 5 1 7
21 21 4 5 15
22 22 1 5 4
23 23 14 5 15
24 24 5 4 13
25 25 3 2 9
26 26 6 4 13
27 27 4 5 14
28 28 4 3 12
29 29 7 1 8
30 30 3 4 5
31 31 3 2 10
Dihedrals
1 1 4 3 2 1
2 2 5 1 2 3
3 3 16 6 4 3
4 4 3 4 5 1
5 5 7 1 5 4
6 6 11 3 2 9
7 7 6 4 5 1
8 8 12 3 2 10
9 9 9 2 1 5
10 10 14 5 1 7
11 11 14 5 1 2
12 12 10 2 1 5
13 13 13 4 5 1
14 14 7 1 2 3
15 15 16 6 4 13
16 16 15 5 1 7
17 17 6 4 3 2
18 18 14 5 1 8
19 19 16 6 4 5
20 20 8 1 5 4
21 21 11 3 4 5
22 22 11 3 2 1
23 23 14 5 4 13
24 24 13 4 3 11
25 25 12 3 2 1
26 26 9 2 3 4
27 27 13 4 3 12
28 28 12 3 4 6
29 29 11 3 2 10
30 30 12 3 4 5
31 31 11 3 4 6
32 32 4 5 1 2
33 33 15 5 4 3
34 34 10 2 1 7
35 35 5 4 3 2
36 36 8 1 2 3
37 37 9 2 1 7
38 38 15 5 4 13
39 39 15 5 1 2
40 40 15 5 1 8
41 41 13 4 3 2
42 42 10 2 1 8
43 43 9 2 1 8
44 44 14 5 4 6
45 45 15 5 4 6
46 46 12 3 2 9
47 47 10 2 3 4
48 48 14 5 4 3
Impropers
1 1 4 3 5 6
2 2 1 2 5 7
3 3 1 2 5 8
4 4 2 9 3 1
5 5 2 1 10 3
6 6 3 2 11 4
7 7 3 4 2 12
8 8 4 5 3 13
9 9 5 1 4 14
10 10 5 1 4 15
