Hello, group members.
I have been experiencing several errors while performing calculations using the API recently. First of all, I am conducting DFT calculations using Quantum ESPRESSO.
Some materials output errors such as lone vector and not orthogonal. Additionally, even for structures that have completed calculations, performing interpolation using BoltzTraP2 results in interpolation being impossible.
In particular, the error that occurs when using BoltzTraP2 is related to irreducible k-points and is an error in failing to properly find symmetry. Of course, I considered that it might be an issue with QE itself, but after looking at the input files, I realized there was another problem.
- The atomic structure .cif files downloaded from the Materials Project site and the .cif files of the primitive structure downloaded using the get_primitive option from the API are very different in structure.
- In particular, if I do not use the get_primitive option and simply output the structure obtained from structure_summary, the output structure is strange.
3.The structure obtained from to_conventional() is more similar to the structure provided by the Materials Project compared to the structure obtained from get_primitive.
Could you let me know how get_structure, get_primitive_structure, and to_conventional are connected in structure_summary, where the structure is sourced from, and how it is transformed?
I would appreciate it if you could let me know what might be causing the above questions. I have added a few example files below.
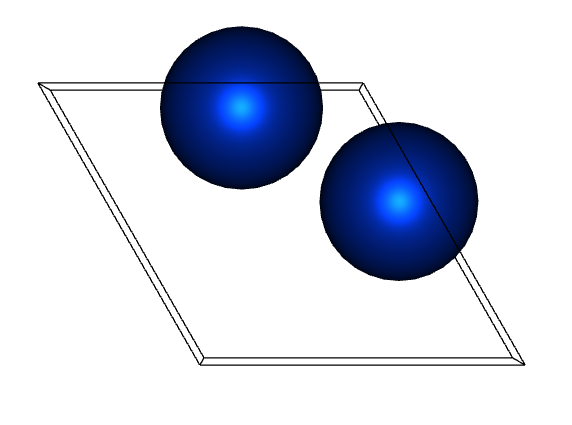
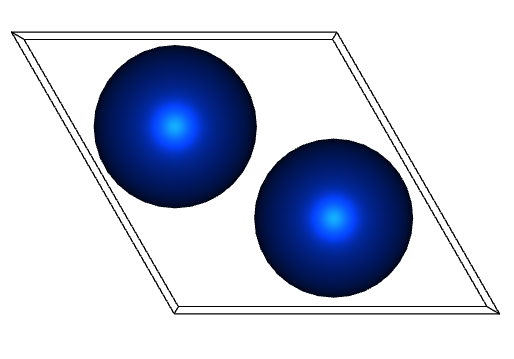
If you look closely, you can see that the atomic position and gamma angle between the two are different.
- Be_mp-87.cif from Materials project
Be from MP
# generated using pymatgen
data_Be
_symmetry_space_group_name_H-M 'P 1'
_cell_length_a 2.26478265
_cell_length_b 2.26478265
_cell_length_c 3.57410246
_cell_angle_alpha 90.00000000
_cell_angle_beta 90.00000000
_cell_angle_gamma 120.00000000
_symmetry_Int_Tables_number 1
_chemical_formula_structural Be
_chemical_formula_sum Be2
_cell_volume 15.87635094
_cell_formula_units_Z 2
loop_
_symmetry_equiv_pos_site_id
_symmetry_equiv_pos_as_xyz
1 'x, y, z'
loop_
_atom_type_symbol
_atom_type_oxidation_number
Be0+ 0.0
loop_
_atom_site_type_symbol
_atom_site_label
_atom_site_symmetry_multiplicity
_atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_z
_atom_site_occupancy
Be0+ Be0 1 0.33333333 0.66666667 0.75000000 1
Be0+ Be1 1 0.66666667 0.33333333 0.25000000 1
- Be_mp-87.cif from API, get_primitive
Be by API, Primitive
# generated using pymatgen
data_Be
_symmetry_space_group_name_H-M 'P 1'
_cell_length_a 2.26478265
_cell_length_b 2.26478265
_cell_length_c 3.57410246
_cell_angle_alpha 90.00000000
_cell_angle_beta 90.00000000
_cell_angle_gamma 119.86323685
_symmetry_Int_Tables_number 1
_chemical_formula_structural Be
_chemical_formula_sum Be2
_cell_volume 15.89818514
_cell_formula_units_Z 2
loop_
_symmetry_equiv_pos_site_id
_symmetry_equiv_pos_as_xyz
1 'x, y, z'
loop_
_atom_site_type_symbol
_atom_site_label
_atom_site_symmetry_multiplicity
_atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_z
_atom_site_occupancy
Be Be0 1 0.58304792 0.91695208 0.75000000 1
Be Be1 1 0.91695208 0.58304792 0.25000000 1
In addition, the results obtained using the to_conventional command are most similar to the results from the .cif file downloaded from MP. What is the criterion for conventional? Where does this value come from?
Be by API, conventional
generated using pymatgen
data_Be
_symmetry_space_group_name_H-M ‘P 1’
_cell_length_a 2.26478265
_cell_length_b 2.26478265
_cell_length_c 3.57410246
_cell_angle_alpha 90.00000000
_cell_angle_beta 90.00000000
_cell_angle_gamma 120.00000000
_symmetry_Int_Tables_number 1
_chemical_formula_structural Be
_chemical_formula_sum Be2
_cell_volume 15.87635094
cell_formula_units_Z 2
loop
_symmetry_equiv_pos_site_id
symmetry_equiv_pos_as_xyz
1 ‘x, y, z’
loop
_atom_site_type_symbol
_atom_site_label
_atom_site_symmetry_multiplicity
_atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_z
_atom_site_occupancy
Be Be0 1 0.33333333 0.66666667 0.75000000 1
Be Be1 1 0.66666667 0.33333333 0.25000000 1
*** My API codes below,**
import os
from mp_api.client import MPRester
with MPRester("MY API") as mpr:
structures = mpr.materials.summary.search(
elements=["Be"],
chemsys="*",
energy_above_hull=(0, 0),
is_metal=True,
)
with open("structure.txt", "w") as file:
file.write(f"Number of documents: {len(structures)}\n\n")
if not structures:
file.write("No results found.\n")
else:
for idx, structure_summary in enumerate(structures):
material_id = structure_summary.material_id
formula = structure_summary.formula_pretty
E_Hull = structure_summary.energy_above_hull
# Is_Metal = structure_summary.is_metal
# stable = structure_summary.is_stable
# crystal = structure_summary.structure
sym = structure_summary.symmetry
structure = mpr.get_structure_by_material_id(material_id)
# primitive_cell = structure.get_primitive_structure()
conventional_cell = structure.to_conventional()