Hi everyone,
I’ve simulated a substrate of Ni atoms with Cu powders on it, and equilibrated the system in 300k with periodic boundraries; the atoms of Ni, starts going on the top of Cu powders,
To solve this problem, I used some force fixes to stop the substrate atoms from moving;
but its temperature changes and wouldn’t be 300k.
I want the temperature of both substrate and powders to be constant on 300k and also stop the substrate atoms from moving, Can you help me with this?
*substrate consists of 4 layers of Ni atoms/
You have to provide some more information about the details of your specific setup and input.
How should that be possible that the substrate atoms can more “on top” of the deposited atoms? Where do they come from? Could it be that your box dimensions are insufficient.
I am also a bit confused about the term “Cu powders” as typical sizes of grains in a regular powder are larger than the entire simulation cell of an atomic-scale MD simulation.
Please note that a typical setup for a substrate would be to have 1-2 immobile layers at the bottom with bulk lattice positions for the given temperature. Then 3-4 layers of mobile atoms that are thermostatted, by preference with a dissipative thermostat (e.g. fix langevin) and then a remaining set of “free” atoms that are initially equilibrated with a thermostat, but then for production calculation are run without a thermostat. Those should then maintain the equilibrated temperature (if they heat up, then there may be issues with other simulation settings like too noisy potentials, or too large a time step or similar).
Actually the powders put pressure on the substrate and cause its atom’s to move then because of the periodic boundary, they appear on the top of powders (Powders and substrates are tangent to the grid dimensions).
I attached the picture if you want to see more accurate.
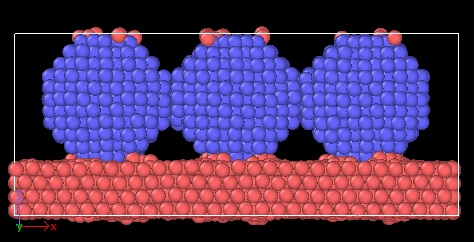
yes, that is almost what I suspected as the worst possible case.
- you should have the setup that I described (and thus have plenty more atoms in the substrate to avoid finite size effects there from the reconstruction and deformation and you must have the immobile bottom layer(s))
- you are not simulating a substrate with deposited nanoparticles but a “sandwich”, due to the use of periodic boundary conditions. You need to have a extra vacuum in the z direction and use fixed boundaries in z not periodic. Then atoms from the bottom do not interact with the top of the nanoparticle.
Ok, thank you for responsing;
just another question, how can I have immobile layers? should I use an specific fix command?
Please note that this is a frequently mentioned topic, so you could have searched the existing discussions before asking. The simplest way to keep atoms from moving is by not including them in the group(s) used for the time integration fix(es). The fix (e.g. fix nve, or fix nvt) will update the positions of the atoms in its group. If atoms are not included in the group, their positions won’t change.
I’ll check, thank you so much.