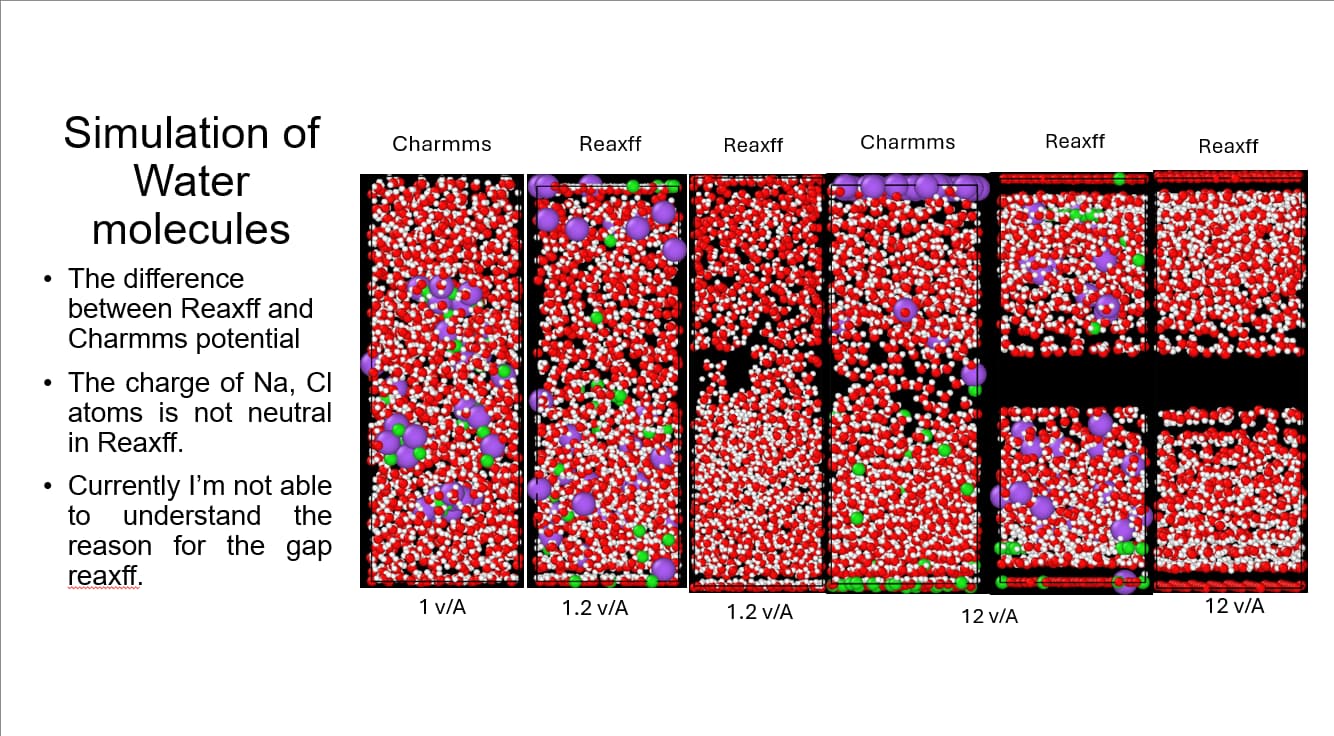
I have a question for all reaxff users and researchers. I’m currently working on the effect of efield on the NaCl+H2O system. I have found the behavior of the system with charmms is much more favorable as compared to behavior with reaxff. Why is there such a huge difference? And why there is split in the system even though the electric field is applied in the complete simulation box. How can I fix this? Is there something that needs to be done in addition to applying efield command?
It will be really great to have an insight on this. Thank you all in advance.
Yes, I do know that I’m applying extreme high electric field, but for understanding the motion of water molecules. It was my understanding that applying efield in positive direction will lead to movement of positive ions towards positive z direction. Am I wrong to expect it? I’m sorry for the way I interpreted my question, what I meant was the combine charge of Na+ Cl- ions should had been zero. But it is not the case.
Sure, positive ions move in the direction of the electric field, but I am not sure how it is related to my comment about the magnitude of the electric field. All I see is that you are applying a crazy value for the electric field, and you observe a crazy behavior.
I’m far from being an expert in ReaxFF but I do know such a model minimizes charges on atoms from time to time. Thus I would check if q_Na + q_Cl → 0 on average. I honestly don’t know if there is any constraint for NaCl being neutral on every timestep.
Thank you for the explanation. I’m sorry, I couldn’t answer your question. So I have applied reasonable value of efield as well which is 0.3v/A, but couldn’t see any change in behavior. I’m working on the NaCl+Water system to understand the behavior of water molecules under efield. This is to know, why the water molecules are moving against the gravity in the below attached simulation. in.FeNiCr.lmp (3.6 KB)
It will do so, but with a reasonable electric field it can take a long time (and it is supposed to take a long time). What you seem to expect is what is shown in computer animations of macroscopic projects. On the atomic scale things are far more subtle and require long trajectories and lots of averaging to “see” anything.
At a crazy strong electric field you are beyond any reasonable physics and thus expecting reasonable results is a folly.
Charges are fixed in CHARMM, but flexible in ReaxFF (with impact of the external potential).
That is very different physics at work.
Please note that a meaningful external field (where the system under investigation would not explode instantly due to electrolysis and sparks causing an explosion of the hydrogen/oxygen mixture) is several orders of magnitude lower. At that point processes are so slow, you cannot afford to run simulations long enough.
Thank you for the reference. I have now a fair idea about it. My system works more with water and metal interactions at the interface. Is there a way to combine the long range coulomb interaction from the Qeq for metals and short range interactions from ACKS2 to avoid over-estimation of charges for interface study under external electric field?
ACKS2 is superior to QEq, no need to combine the two. ACKS2 is valid for metals too. As implemented in LAMMPS neither QEq nor ACKS2 handle long-range coulomb correctly. This is because ReaxFF truncates the coulomb interaction with a damped cutoff like a Wolf summation, instead of using an Ewald sum or PPPM. This is OK when there is charge screening, but maybe not for an interface. The problem with ACKS2 is you likely need to reparameterize the entire ReaxFF potential. You could also look into QTPIE:
Can I use the attached reaxff potential parameter file with ACKS2?
Unless the reaxff potential parameter file was fit with ACKS2 (originally) then probably not. ACKS2 is relatively new so unfortunately there aren’t many force fields that support it. Both ACKS2 and QTPIE require additional parameters beyond QEq. I think QEq to QTPIE is a little more transferable since you can recover QEq behavior if you make certain parameters zero for QTPIE (or something like that). You could check with Adri van Duin, who would know best if there are any new FFs that support ACKS2.
But the QTPIE requires Gauss Exponents, which are not available at current moment. Is it possible to run the simulation making those values 0 or can it be done without the Exponents file?
I think it would revert back to QEq without the extra parameters, which defeats the purpose of using QTPIE in the first place. This is all bleeding edge research so unfortunately there isn’t a “turnkey” solution available, at least that I know of…
Thank you so much for all the information that you have provided. Would it be alright to use the orbital exponents value from the experimental literature?