Dear lammps-users,
I was trying to use TIP4P pair style in lammps to realize OPC water model proposed in
https://pubs.acs.org/doi/10.1021/jz501780a
I changed the parameters to
pair_style lj/long/tip4p/long long long 1 2 1 1 0.1594 8.0
bond_style harmonic
angle_style harmonic
kspace_style pppm/disp/tip4p 1.0e-5
kspace_modify force/disp/real 0.0001
kspace_modify force/disp/kspace 0.002
…
pair_coeff * * 0 0
pair_coeff 1 1 0.22259 3.16655
…
Bond Coeffs # harmonic
1 0.0000 0.8724 # o-h
Angle Coeffs # harmonic
1 0.0000 103.600 # h-o-h
…
fix 1 all shake 1.0e-4 200 0 b 1 a 1
fix 2 all npt temp 298.0 298.0 100.0 iso 1.0 1.0 100.0
Data files were obtained by Materials studio with the corresponding bond length and angle. I am not sure whether it is correct to use lj/long/tip4p/long to achieve both long range Coulombic interaction and dispersion correction emphasized in most papers concerning water model. As I tried, using lj/long/tip4p/long obtained larger and more reasonable density (~0.996) of water than lj/cut/tip4p/long (~0.98).
Then I used following method to compute the fluctuations of total dipole moment
compute 11 all property/atom xu yu zu q
variable dmx atom c_11[1]*c_11[4]
variable dmz atom c_11[2]*c_11[4]
variable dmy atom c_11[3]*c_11[4]
compute 12 all reduce sum v_dmx v_dmy v_dmz
variable dm2 equal c_12[1]^2+c_12[2]^2+c_12[3]^2
variable vvol equal vol
fix fdm all ave/time 1 10000 10000 c_12[1] c_12[2] c_12[3] v_dm2 v_vvol file dipole.csv mode scalar
Dielectric constant was calculated by
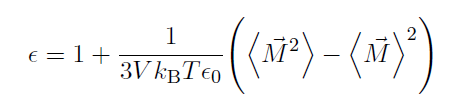
But the final value remained stable around 158 after running for 20ns, which is almost twice the expected value. The simulation was conducted on windows server 2016 with lammps version of 8April2021.
Is there any problem in my simulation, about modelling and dielectric constant computation? Hope to get your help.
Regards,
Zhang SW
1 Like
I cannot comment on the details of your calculation(s). The determination of the static dielectric constant is a complex task, so it is possible that things go wrong in all kinds of places and it is usually not possible to verify the results from a few extracts of your input file; it would take significant time and access to suitable facilities to re-run the calculations independently.
I have done a survey of available results and did some comparisons of different models, finite size effects and how to do the necessary calculations a long time ago, which are summarized in the attached PDF. Perhaps you can find something useful in there.
talk-trieste2004.pdf (1.1 MB)
Hi Axel,
Thanks for your reply. My previous simulation box contained 2000 water molecules. Just now I tried with less (64) water molecules and different parameters such as timestep, damping factor of thermo- or barostat, cutoff value. I still got similar dielectric constant for OPC water about 150+. However, the dielectric constant of SPC/E model I realized seems to be correct.
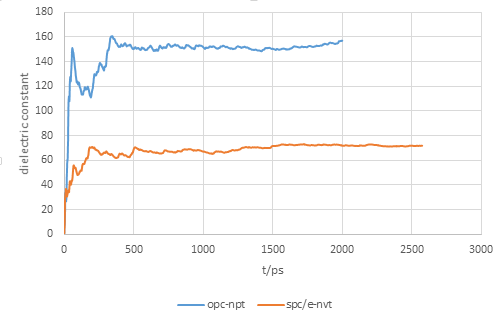
I also compared my result with the python script published in
https://pubs.acs.org/doi/10.1021/acs.jcim.9b00066
So I believe that my calculation of dielectric constant should be reasonable. I wonder that when calculating the dipole moment in OPC model or TIP4P model, should we consider the coordinate of O site of water or the massless site M? It seems that my current method considers the position of O site but not the massless site where actually the charge is placed.
I have attached my input file and data file.
Zhang SW
in.opc.lmp (1.6 KB) in.spce.lmp (1.6 KB) water-opc64.data (23.5 KB) water-spc.data (23.5 KB)
1 Like
This is a very good question. Indeed, you must not use the location of the O point for computing the dipole moment of the water molecules. You can easily check this by computing the dipole moment of individual water molecules. I should be significantly larger than the expected value if you use the oxygen atom location. When you apply the charge to the point M (which you have to compute on the fly) then it should be correct.
I think for the corresponding computations from inside LAMMPS, there needs to be a check for the presence of a tip4p pair style and then a warning printed that the dipole moment will not be correct.
Hi Axel,
The problem needs to be paid attention and I found that someone put similar question in mailing list, only after I had asked the question.
https://sourceforge.net/p/lammps/mailman/message/34812187/
Anyway, I have tried to use compute and variable command to get the dielectric constant for TIP4P pair style. The vector between O site and M site is related to H site vector.
compute 11 h property/atom xu yu zu
compute 10 o property/atom xu yu zu
variable phi equal 0.14772952 #OMdistance/(OHlength*cos(HOHangle/2)*2)
variable dmx atom 0.6791*c_11[1]-1.3582*(c_10[1]*(1-2*v_phi)+c_11[1]*v_phi)
variable dmz atom 0.6791*c_11[2]-1.3582*(c_10[2]*(1-2*v_phi)+c_11[2]*v_phi)
variable dmy atom 0.6791*c_11[3]-1.3582*(c_10[3]*(1-2*v_phi)+c_11[3]*v_phi)
compute 12 all reduce sum v_dmx v_dmy v_dmz
variable dm2 equal c_12[1]^2+c_12[2]^2+c_12[3]^2
variable vvol equal vol
variable vrou equal density
fix fdm all ave/time 1 1 1 c_12[1] c_12[2] c_12[3] v_dm2 v_vvol v_vrou mode scalar ave running
variable dc equal 1+(f_fdm[4]-f_fdm[1]^2-f_fdm[2]^2-f_fdm[3]^2)*(1.6022e-19*1e-10)^2/3.0/f_fdm[5]/1e-30/1.38065e-23/298/8.85418e-12
Now I can obtain result closer to expected value. Thanks again!
Zhang SW