Hello all. I want to establish a composite model of oil and water. First, I ran the model of oil in the NPT ensemble for 1 ns using the PPP boundary. However when I used read_data command to combine the model of oil and water, due to the presence of periodic boundaries, the bonds between molecules in the model of oil were elongated. I wonder how to solve this problem. Thanks a lot.
combine.in (392 Bytes)
final.data (493.4 KB)
water.data (156.0 KB)
oil-water.data (732.6 KB)
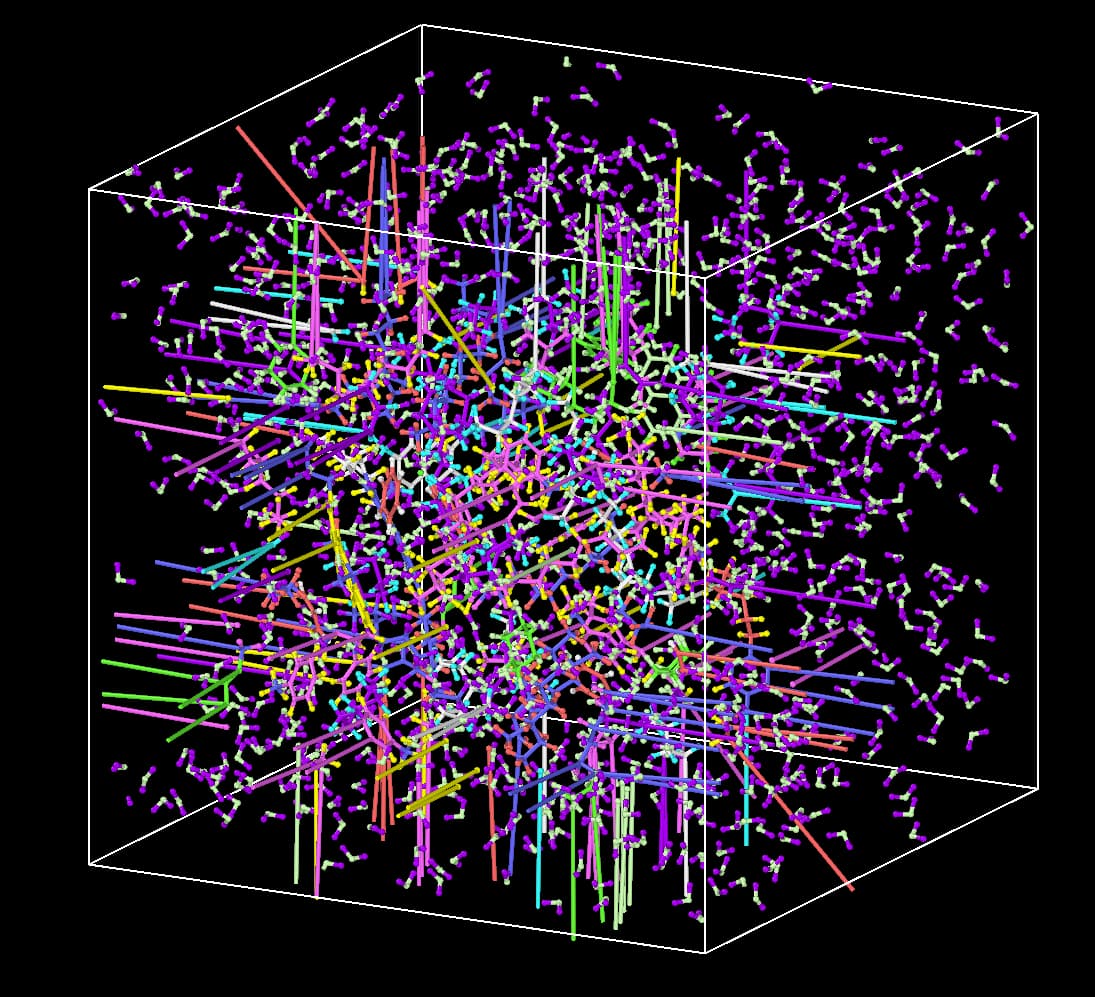
If you are concerned about the effects of the periodic boundary, have you checked if you are reading in wrapped or unwrapped coordinates from your data file?
Sorry, I didn’t understand what you meant. I know this problem was caused by atoms exceeding the boundary after NPT. I just whether how to ensure that no atoms exceed the boundary while operating in an NPT ensemble under the premise of using pppm to calculate long-range forces.
It is normal for atoms to travel outside the boundaries, given that a long-range solver requires periodic boundaries. I am not sure if the current configuration can cause problems because you have molecules effectively split between different domains.
I have loaded your data file in VMD, but it fails to unwrap the molecules:
topo readlammpsdata oil-water.data
pbc join res
pbc unwrap -now
Sometimes pbc wrap -compound resid does the trick, but not in this case. @baerb is suggesting to dump your system with unwrapped coordinates if you want to avoid this kind of problems, e.g. using
dump TRJ all custom 100 test.dump id type xu yu zu mol
instead of ... x y z ....
So if each model has operated under an NPT ensemble before being combined, how can I combine the models together?
Oh, now I see the problem.
When you combine two data files together, the resulting box is taken from the larger one. In your case, the water box is the largest. The problem is that the image flags of the first box will now be applied with different box vectors, leading to split molecules. A workaround is to remap the coordinates of the first data file, which will use the image flags correctly, but will also stretch the atom positions to unphysical values.
combine1.in
# Define the force field.
units real
atom_style full
bond_style harmonic
angle_style harmonic
dihedral_style harmonic
improper_style umbrella
pair_style lj/cut/coul/long 10
kspace_style ewald 1.0e-4
# Read and combine two systems.
read_data final.data extra/atom/types 2 extra/bond/types 1 extra/angle/types 1
change_box all x final 0 45 y final 0 45 z final 0 45 remap
read_data water.data add append offset 50 69 30 45 11 shift 0 0 0
# Write the unwrapped coordinates.
dump TRJ all custom 1 oil-water.dump id type xu yu zu mol
run 0 post no # That's for the DUMP.
write_data oil-water.data
A better way to proceed is to read the two systems independently and save the unwrapped coordinates in separate DUMP files:
combine2.in
# Define the force field.
units real
atom_style full
bond_style harmonic
angle_style harmonic
dihedral_style harmonic
improper_style umbrella
pair_style lj/cut/coul/long 10
kspace_style ewald 1.0e-4
# Read the first system.
read_data final.data
# Write the unwrapped coordinates.
dump TRJ all custom 1 final.dump id type xu yu zu mol
dump_modify TRJ sort id
run 0 post no
undump TRJ
# Define the force field.
clear
units real
atom_style full
bond_style harmonic
angle_style harmonic
dihedral_style harmonic
improper_style umbrella
pair_style lj/cut/coul/long 10
kspace_style ewald 1.0e-4
# Read the second system.
read_data water.data
# Write the unwrapped coordinates.
dump TRJ all custom 1 water.dump id type xu yu zu mol
dump_modify TRJ sort id
run 0
From this, you obtain two dump files that can be combined into a single one, which you can use to replace the coordinates in the original DATA file. In this case, you have to get rid of the image flags.
Here is the sample after merging the two files:
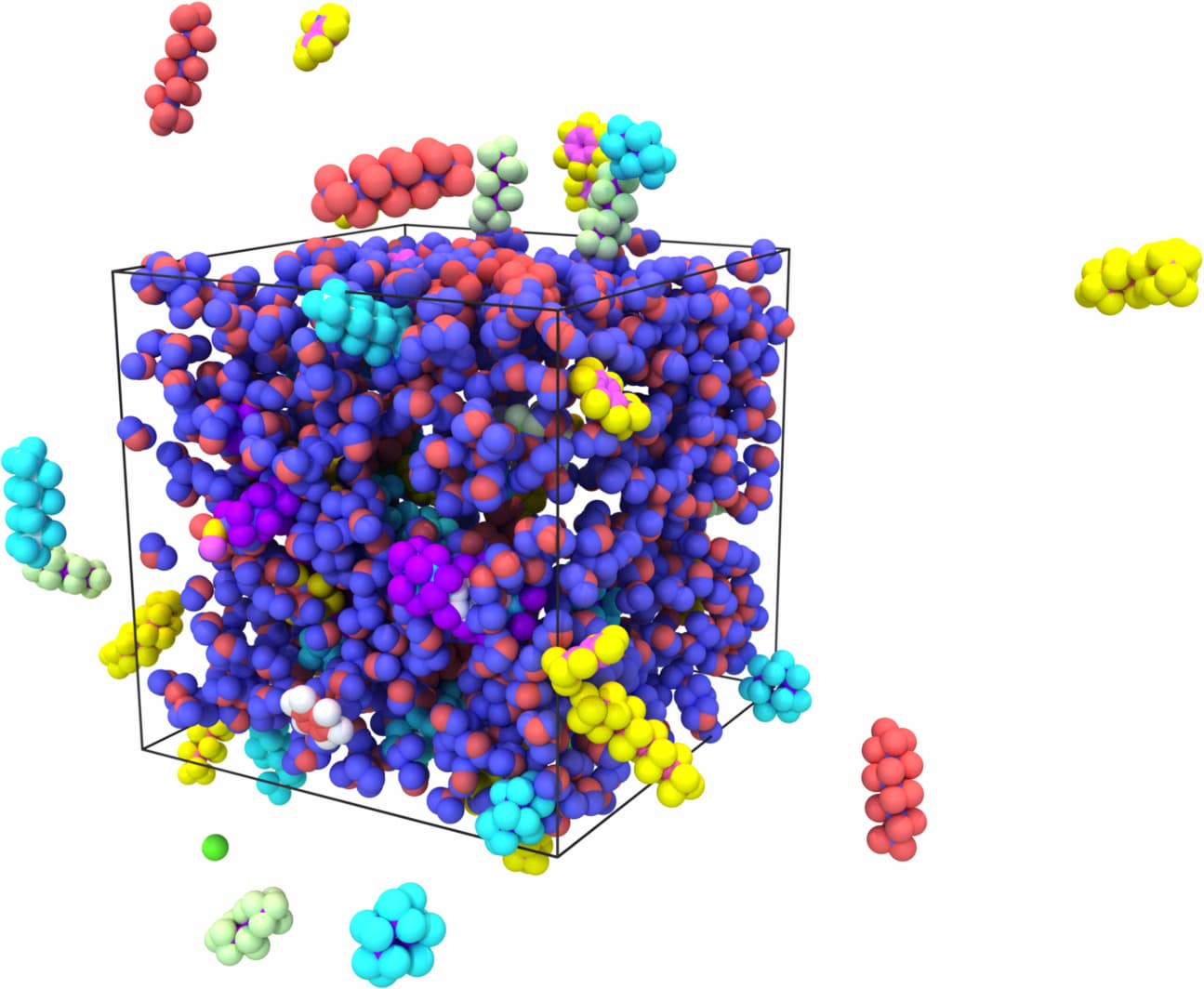
I would go for the latter approach to avoid working with a very deformed initial structure. To give you an idea, this is a comparison of the bond lengths for resid == 60:
| combine1.in | combine2.in |
|---|---|
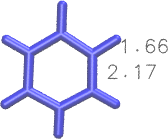 |
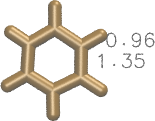 |
Maybe this issue could be solved with a new option for read_data that unwraps the coordinates after reading them. Or maybe we are pushing LAMMPS too much outside its legitimate usage.
It is a combination of that, the person implementing this feature not caring enough (it worked for him), and the read_data code being a bit of a mess so any attempts to make things more general will trigger many possible unexpected problems.
The best strategy for me has always been to adjust the box for each of the component subsystems before attempting to merge them, so there is no more problem with image flags. This can be done with something like the following (this is off the top of my head and untested):
reset_atoms image all
group none empty
change_box none x final ${xlo} ${xhi} y final ${ylo} ${yhi} z final ${zlo} ${zhi} remap
write_data final_box.data pair ij
The first command will reset the image flags to that the closest images are used.
Then the empty group is needed so that change_box won’t scale any atom positions and the remap option should recompute the atom coordinates so that the image flags now match the new box.
At least that is the idea. For details one has to look into the documentation and make some experiments with some simple system to confirm.
Personally, I usually do the merging process in VMD. But that is because I am very familiar with it.
Yeah, I also prefer to do the merging outside LAMMPS from DUMP files with unwrapped coordinates. I have added a DUMP parser to Moltemplate to make it easy to assemble larger systems and then load them back into LAMMPS with a properly formatted DATA file.
Update
The change_box command applied to an empty group does not fix the coordinates of the original data structure.
combine3.in
# Define the force field.
units real
atom_style full
bond_style harmonic
angle_style harmonic
dihedral_style harmonic
improper_style umbrella
pair_style lj/cut/coul/long 10
kspace_style ewald 1.0e-4
# Read the first sample
read_data final.data extra/atom/types 2 extra/bond/types 1 extra/angle/types 1
# Remap the atoms to the largest box (use random numbers for missing coeffs).
pair_coeff * * 1 1 10
bond_coeff * 2 2
angle_coeff * 2 2
mass 51*52 10
reset_atoms image all
group none empty
change_box none x final 0 45 y final 0 45 z final 0 45 remap
# Add the second sample.
read_data water.data add append offset 50 69 30 45 11 shift 0 0 0
# Write the combined system.
write_data oil-water.data nocoeff
Thanks for your advice, I’ll give it a try.
Nah, you can use LAMMPS to part the Red Sea, combining systems like this is nothing.
Actually, that thread was also about incompatible periodic boundary conditions, and it’s worth a read.
1 Like
I still have some questions. Before I tried this approach to establish model and finally ran a complete simulation successfully. However, I just discovered that if I use the unwrap trajectories, I will find following situation. Although it still can run the simulation normally, it seems to have problems somewhere.
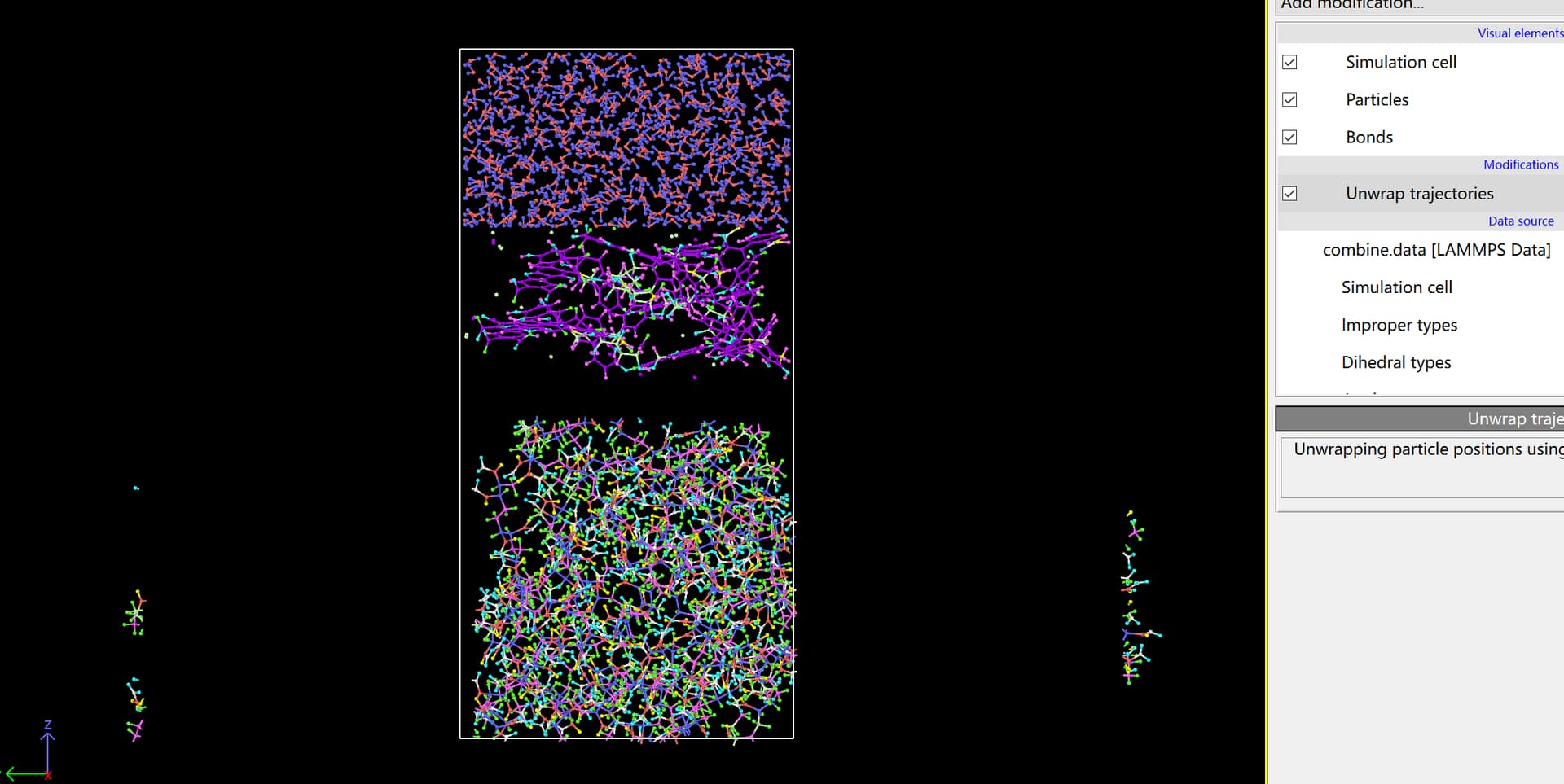
If the simulation runs correctly, the image flags should be correct. Try first to wrap and then unwrap the entire sample, that should heal the broken molecule. There is also try the pbc tools in VMD, for instance with the pbc unwrap and pbc join commands.