I am looking to replicate a similar study by combing a reax/c potential type with a zbl using a hybrid/overlay. When I see similar input files they define pair_coeff for each of the zbl options (including mixing). For example,
pair_coeff 5 2 zbl 18 11
Whenever I specify this mixing I get an error “incorrect args for pair ceofficients”
When I remove the mixing it works. Does zbl automatically account for the mixing?
This is my forcefield definition:
pair_style hybrid/overlay zbl 2 3 reax/c NULL
pair_coeff 1 1 zbl 13 13
pair_coeff 2 2 zbl 11 11
pair_coeff 3 3 zbl 8 8
pair_coeff 4 4 zbl 14 14
pair_coeff 5 5 zbl 18 18
pair_coeff * * reax/c albitereax.ff.txt Al Na O Si O
As a follow up I have been investigating the system after getting the hybrid/overlay to work. I am a noticing a curious development with the temperature of the substrate. Of note, I am loading in an albite substrate from materials project, equilibrating, replicating, equilibrating then reading this surface in. This method has worked in the past with crystallinity maintained and no energy spikes
Summary below:
when reax only is used the substrate equilibrates with no big temp spikes
when hybrid/overlay is used and only the pairs of the same type (1-1, 2-2, etc) are defined, long with the zbl for incoming atom and substrate atoms (1-5, 2-5, 3-5, etc.) everything also works fine with temperatures during equilibration
finally, when I go ahead and define all interactions within the albite (1-2, 1-3, etc.) the substrate temp immediately spikes (up to 2000K) then starts going back down.
This large spike is confusing me and I am not comfortable continuing on until I know why. It makes me feel like I am doing something wrong in zbl. One option is to just define zbl interactions for the incoming projectile and the surface, but either way I would like to know why energies are spiking for atom-atom interactions within the albite.
My reax definition is:
---- potential ----
pair_style hybrid/overlay zbl 2 3 reax/c NULL
pair_coeff * * reax/c albitereax.ff.txt Al Na O Si O
pair_coeff 1 1 zbl 13 13
pair_coeff 2 2 zbl 11 11
pair_coeff 3 3 zbl 8 8
pair_coeff 4 4 zbl 14 14
pair_coeff 5 5 zbl 18 18
I don’t have an explanation, but a suggestion: You can compute the pe/atom and then output and visualize it and look at which specific pairs of atoms have the unexpected high energy. Then, you would also monitor how the geometry for those atoms changes as the energy spike dissipates. That should give you some hint of what happens.
Please note that with pair style hybrid/overlay you are adding potential energy to an existing pair style and its parameters. They have repulsion already parameterized (just not well for high impact simulations), so ideally you would need some kind of “switching” between those two potentials instead of just addition.
As per your last point, this is something I have been thinking of myself as well. Maybe in that case I really only need to define the ZBL for the interactions between the incoming projectile and the substrate. Many simulations seem to use a pure zbl for those interactions.
In that case perhaps it makes sense to use a hybrid instead of a hybrid/overlay, and just have pure zbl for interactions between impactor and substrate? I thought about doing hybrid as opposed to hybrid overlay but have read that is not advisable using reax.
Yeah, ReaxFF is tricky business and especially with hybrid models. That is because for correct charge equilibration and automatic bond order calculations you need to consider the interaction of all atoms. However, there would be some justification for your specific case, if you are aware of the implications on the model and make an informed decision if that is a good (enough) approximation and (possibly) better than including everything.
However, if you would be using pair style hybrid and not hybrid/overlay and then apply ReaxFF only to the non-projectile atoms and define pair style zbl only for interactions between substrate and projectile and use pair style none or zero for projectile-projectile interactions and also define a group for only the substrate atoms and apply fix qeq/reaxff only to those, then this could work. This way your projectile atoms would be basically “invisible” and have no interaction beyond the impact and what is following in the substrate because of that. They are hitting atoms in high speed in a purely “mechanical” fashion without participating in reactions after they have slowed down. In my experience you would need ReaxFF primarily because you what to see the secondary effects on the substrate from the impacts.
In short, instead of double counting as with hybrid/overlay, you are underestimating the interactions with the projectile atoms and reduce them to the initial impact on the substrate.
Agreed, I tend to only use ReaxFF when needed as it also is pretty computationally intense. However for the mineral I am considering it is hard to find another option properly parameterized (I have looked at Teter/Buckingham in the past too but ran into some issues). As you said, with these sputtering simulations I am moreso focused on the result of the impact and cascade on the substrate, and less so on the chemical interactions between the impactor and substrate. I published something similar on Ar impacts on Cu but it was much easier than more complex minerals (Cookie Absent). This study builds on this by considering solar wind interactions with a lunar material.
As I think more I think hybrid (as opposed to overlay) may be my best way forward. Then ZBL only for substrate projectile as you said. That way I don’t have any “odd” reaxFF interactions between projectile and substrate that I may not want.
Also, good point on the qeq/reax using a specific group. I think I would have forgotten that. I’ll keep you posted!
The temperature spike after adding ZBL is an expected behaviour, because you are adding an additional interaction to every pair of atoms which are separated by less than 0.3 nm. The region of the switching function should begin at value lower than the length of the shortest bond in your system. However, you will still have two sources of repulsive force at once (from ZBL and ReaxFF).
I had a similar problem and I solved it by introducing a tabularized potential which was a difference (after a suitable splining region) between ZBL and ReaxFF repulsive forces. Then, I used it in place of ZBL, to get a pure ZBL force at short distances. You can find a better description in my paper: https://doi.org/10.1021/acs.jpclett.7b03155
Splining ReaxFF and ZBL can be tricky, especially if repulsive barriers in ReaxFF parametrization are low, so you should always confirm that the transition from one to another is smooth.
Hi Michal, thanks for sharing. I am actually familiar with that paper, in an ideal world reax potentials would have this zbl added in during parameterization.
I agree that because I am adding in a zbl there is now additional energy. I just did not anticipate such a spike. It makes me wonder if reax was already doing an adequate job before. I don’t think there should be large energy spikes given the initial set up of the albite (nothing should be too close approached)
A couple of questions to your post:
I have always been a little confused about proper selection for ZBL rinner and router cutoffs. If ZBL defines the high energy repulsive energy at close approach, then at far approach wouldn’t it just go to close to zero automatically? In other words, even if my switching function began very far away, wouldn’t the zbl contribution already be very low at this point? It seems difficult to pick a good ZBL rinner and outer because you can only define one set for the whole system. When I did into some tersoff/zbl combinations I seem to regularly see switching beginning at 2 and going to zero at 3.
as a follow up to q1, is this why you are recommending that zbl switching begins within the smallest bond, to basically make sure it doesn’t add excessive energy to interactions that are actually not “close approaches” but instead are crystalline arrangements?
As an alternative, I am leaning towards avoiding the overlay all together and have purely repulsive defined for interactions of impactor and substrate.
Thanks for the discussion, always nice to talk to people a little more involved in the parameterization side of things.
Most of ReaxFF parametrizations were created for much lower kinetic energies (tens of eV at most), so ZBL wasn’t really necessary. There are some ReaxFF potentials (like ReaxFF-lg) which have better repulsive barriers, but they are still far from ZBL. I agree that having it incorporated into ReaxFF would be the best solution.
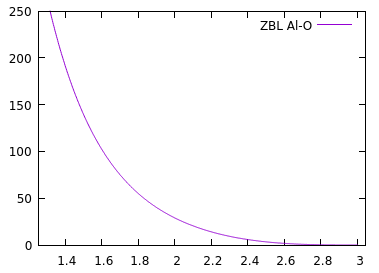
Keep in mind that ZBL is purely repulsive and it was designed for high kinetic energy impacts (at least few keVs), so it’s pretty strong and it doesn’t take into account that atoms can bond with each other. The figure below shows ZBL energy curve for Al-O interactions (y-axis is in kcal/mol, x-axis in angstroms) with the switching region from 3A to 2A.
Assuming the Al-O bond length in your structure is 1.8A, you add about 50 kcal/mol of energy per bond, which leads to the large energy spike.
This is why the splining/switching region has to start at value lower than the shortest bond. You can circumvent the limitation of a single pair of r_inner and r_outer by tabularizing the interactions beforehand (with different switching regions) and using pair_style table instead of pair_style zbl.
Limiting impactor-sample interactions to ZBL may be acceptable, especially if the kinetic energy is high enough. I recall reading a paper about the applicability of ZBL to low kinetic energy impacts, but I can’t find it now.
There is also one additional issue you haven’t considered - first few collisions will create substrate atoms with high kinetic energy, but they will be interacting with the rest of the sample only through ReaxFF. You should think about whether this is enough or if ZBL should be used here also. It all depends on the primary kinetic energy.
LAMMPS code for creating the data for the figure:
atom_style full
units real
region u sphere 0 0 0 30
create_box 2 u
mass 1 1
mass 2 1
pair_style zbl 2 3
pair_coeff 1 1 8 8
pair_coeff 2 2 13 13
pair_write 1 2 1000 r 0.1 3.0 table.txt test
Finally got around to working on this again (bit of a passion side project). Was going to make a new post but seems to make more sense to keep it in the current thread.
Because I am only concerned with collisional Ar impacting the albite I decided to use a hybrid of ZBL and Reax. Reax for albite, ZBL only for interactions between the projectile and any albite atom.
I have used this approach with success before for a tersoff and EAM style potential. When using it with reax I would get random massive energy spikes leading to the simulation box exploding. I tried several things to source the error including:
a different reax potential
a different incoming atom
smaller timestep
larger simulation box
removing layers on the side at a fixed temp
What I ended up finally determining is that the qeq/reax seems to be causing the issues. When I make checkqeq no everything works rather nicely. Is there a reason why for this sort of hybrid style qeq/reax would cause issues?
I should add that I also tried running a qeq/reax on atom types 1-4 only but still ran into the issue.
Likely as you said trying to do charge equilibration when a random atom is stuck in the middle is probably the problem. I did think though that running qeq/reax on atoms 1-4 only would have worked.
why when I do the qeq/reax to albite atoms only (type 1-4) do I still get the explosion?
group albite type 1 2 3 4
pair_style hybrid zbl 3 4 reax/c NULL checkqeq no
pair_coeff 5 5 zbl 18 18
pair_coeff 1 5 zbl 13 18
pair_coeff 2 5 zbl 11 18
pair_coeff 3 5 zbl 8 18
pair_coeff 4 5 zbl 14 18
pair_coeff * * reax/c psof.ff Al Na O Si NULL
fix 21 albite qeq/reax 1 0.0 10.0 1.0e-6 reax/c
If I am only concerned with the impact of the Ar on the albite, will turning off charge equilibration cause an issue to the bonding or realism of the simulation?
pair_coeff * * reax/c albitereax.ff.txt Al Na O Si O
fix 21 all qeq/reax 1 0.0 10.0 1.0e-6 reax/c
I have also tried these using hybrid instead of hybrid overlay
In all cases I am getting convergence issues and a random massive spike in temperature that can only be resolved by turning off qeq/reax. I think qeq/reax is needed to have proper energy in the system so I am really trying to source the issue here.
There is too much marginal detail, too much speculation, and too little systematic narrowing down of the root issues. I don’t have the time to extract the relevant information from this. I suggest you produce a more minimal example to address the various issues one by one. In reference to charge equilibrartion I should point out that you are not locked into using fix qeq/reaxff, you can also use fix qeq/shielded which is a different implementation of the same algorithm as part of the QEQ package.
Some ReaxFF parametrizations are just unsuitable for bombardment simulations. Their parameters governing charge equilibration work properly only if atoms are sufficiently far from each other. If they are too close (tenths of an angstrom) their charges raise sharply leading to the explosion. AFAIK, there’s no solution here other than changing parametrization. I haven’t tried using different charge equilibration implementations though.
Disabling charge equilibration at the beginning (after assigning starting charges) will change bond energies by about 15% (the value differs depending on parametrizations and systems).
@mkanski Thanks for the response. I am thinking this is the issue I am having. This would explain why it is seemingly random (ie not every impact causes the spike), doesn’t go away with reduced time step, happens only at higher energies (600 eV and above), and only will go away if I remove qeq. I also tried it removing the zbl (just a reax impact) and again saw this random massive temperature spike leading to extremely high energy emission.
When you say disable charge equilibration at the beginning, do you mean explicitly defining charges via set charge or by running a qeq in the beginning (before the impact) and then turning it off?