Guys, I am new to LAMMPS could someone help me run the simulation on all processors?
No problem. Just put it in a box and ship it to my office address and I will happily will make certain it is running a simulation on all processors. ![]()
Otherwise there is a lot of useful information in the documentation:
https://docs.lammps.org/Run_head.html
https://docs.lammps.org/Speed.html
Okay, I will have to be specific, the mpich2 software when I tried to install it, flagged me a message stating that .NET version 2.5 was not installed despite me installing it, beforehand. could you suggest something?
I wanted to clarify, This error was flagged despite me installing all the versions of .NET
That indicates that you forgot to mention that your laptop is running windows.
I have never seen that error message but it makes no sense. Which version of MPICH from where were you trying to install.?
Depending on the details of your simulation, you may (after uninstalling those useless .Net packages) install the non-MPI version of LAMMPS and use OpenMP parallization.
I used the MPICH from the LAMMPS Windows Installer Repository.
A quick session with google search reveals, that a) you don’t need to download any specific version of .net explicitly, but rather b) need to enable .Net support in your Windows version. Specifically:
You need to launch the Control Panel and go to where you can configure optional Windows features:
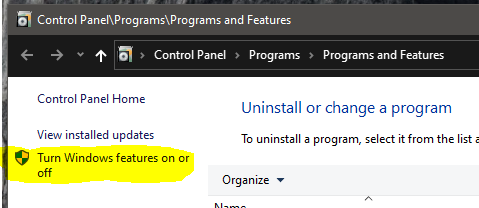
Then you need to enable .Net 3.5 (and older):
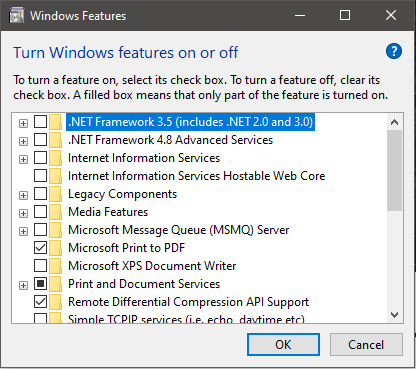
That will prompt you do download updates:
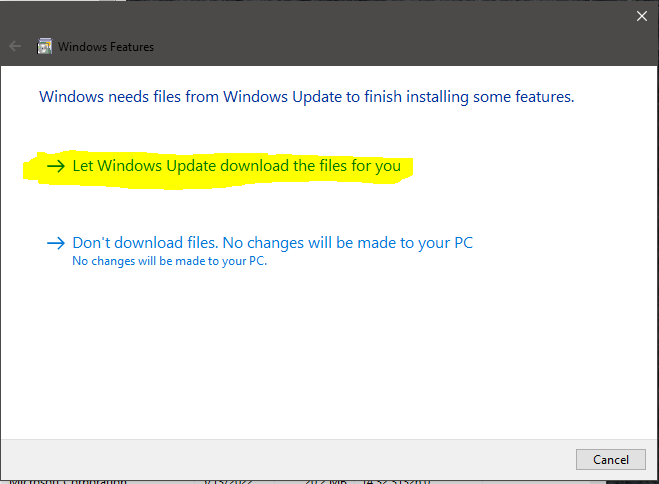
After that is completed, you should be able to install the mpich2 package.
Yes, it worked
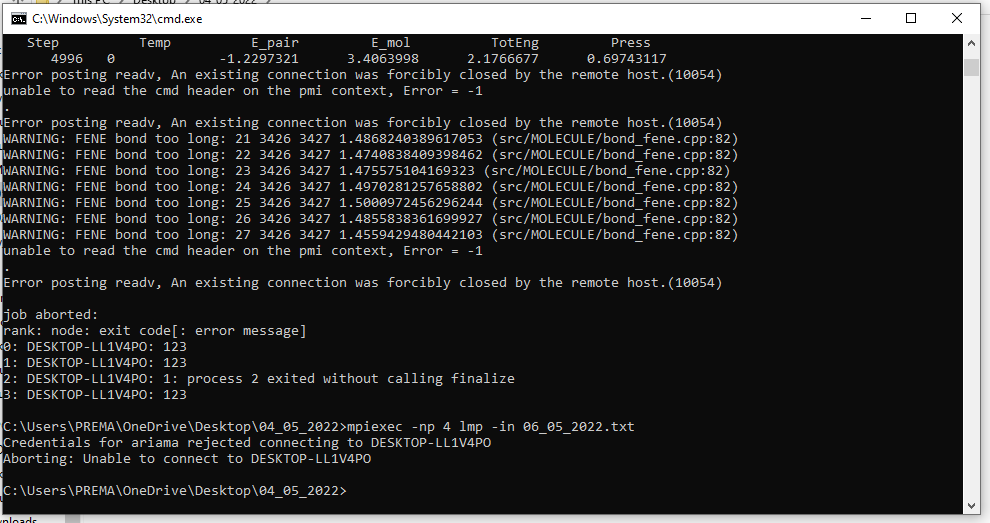
I am getting this error when I use the mpiexec -localonly command. what do you think is the reason? when I switch to other command mpiexec -np it fails.? could you see the problem.
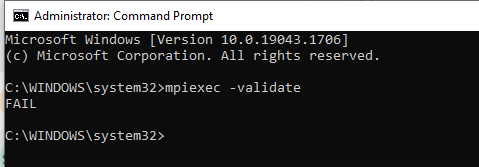
when I tried to validate my credentials it flagged an error like this …in windows? what could be the reason?
See the “WARNING” lines? That is not a good sign. Something seems to be not proper with your geometry, your force field parameters, your simulation settings or a combination of those.
Did you do this?
After the installation of the MPICH software, it needs to be integrated into the system. For this you need to start a Command Prompt in Administrator Mode (right click on the icon and select it). Change into the MPICH2 installation directory, then into the subdirectory
binand executesmpd.exe -install.
and perhaps reboot after that?
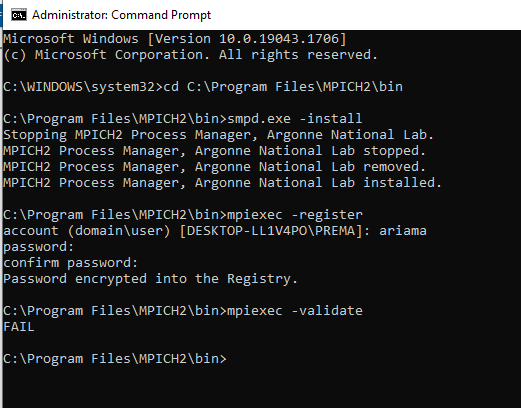
This what I did
That is the exact system I want to simulate…I was able to get the result promptly when running it in just one processor.
Can you run one or more of the provided example input decks? e.g. the in.lj or in.rhodo inputs in the bench folder. If using -localonly is sufficient to run those and the log file correctly reports the number of processors, then LAMMPS is working properly in parallel. To make the -np flag working is not required.
06_05_2022.txt (3.7 KB)
This is the input I am running on 4 processors as of now
The input won’t work without the corresponding molecule file.
I have all the molecule template in the same directory of the input file…
Of your laptop, but they are lacking on mine and thus I cannot try to reproduce your errors on a Linux machine where debugging is infinitely easier than on a Windows machine.
I ran it in one processor, I got bond atoms missing. from the box, I think I will have to make the system much more sparse.