I am a new user of LAMMPS and still learning how to use it.
I am trying to use bonded particle model to simulate the stretching of a beam.
Atoms(337:368) are used to simulate the boundary conditions, so the beam will stretch along x direction. However, I did not get the desired results.
The computed force does not equal to stiffness times change of distance between two atoms.
Did I make some mistakes about bond coefficients?
The version I am using is 23 Jun 2022 version.
I have attached the link for 1) script; 2) molecule; 3) result
Thanks for your response. Hope everyone have a nice day
Hi Stan, would you please clarify what computed force you are seeing on which atom and timestep and how it incorrectly compares to your expectation?
I checked the forces on a few atoms (by dumping fx in dump style custom) and they seem correct at 1 timestep (e.g. forces of 20 with a displacement of 2.5e-08 and a stiffness of 8e8). However, it’s possible that the default force components of compute bond/local (fx fy fz) may be missing a factor of 1/r or something. I will have to look closer at the code to confirm and make sure I’m not misinterpreting the documentation which I may not have a chance to do so until next week. If this is the issue you are seeing, fortunately it does not affect dynamics or reported stresses.
Since the rotational bond style can also transmit tangential forces and compute bond/local returns only the normal force component with fx, fy, fz values it may be more helpful to print the custom b1…bN values which should accurately report the x y and z components of the total force (normal + tangential) as described on the bpm/rotational bond_style page.
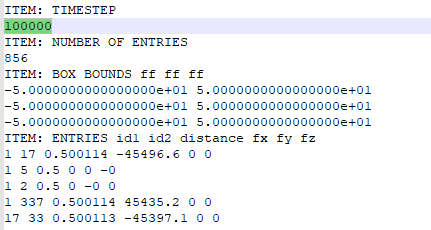
So I use compute bond/local command and check forces at timestep 100000 (you can check the forceall file I uploaded before). For bond between atom1 and atom337, the current distance is 0.500114, so the expected force is
8e8 * (0.500114 - 0.5) = 91200
However, the dump force is 45435.2
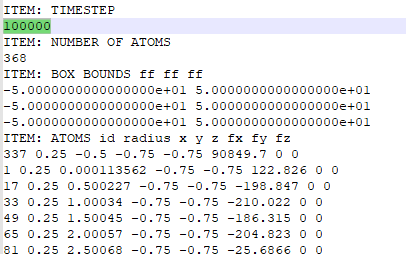
I followed your recommend and use dump style custom to check the force acting on atom337. Fx equals 90849.7, which is close to expected force.
It is kind of weird that the forces are different. Could you tell me the difference between these two command and which one I should use?
That sounds like the value output by compute bond/local may in fact be missing a factor of 1/r. I’ll try to take a look next week and confirm.
Bond styles have two methods which calculate forces: compute() and single(). The compute() method is the primary method which calculates forces on all bonds and applied to each atom. The single() method is an optional method which (I think?) is only really used by compute bond/local to calculate the force on a single bond. It does not affect atom trajectories. The output of the two methods are probably different simply because the single() method mistakenly has an extra factor of r.
Since bond style bpm/rotational is an odd bond style which calculates tangential forces, the documentation for compute bond/local may also not accurately reflect that the output for “compute 1 all bond/local fx fy fz” will actually only return the x, y, z components of the normal force (currently w/ an extra factor of r) while “compute 1 all bond/local b4 b5 b6” will actually return the correct x y z components of the total force (normal + tangential).