I’m running simulations consisting of ~2000 molecules using LAMMPS version 23 June 2022 and ReaxFF as the force field.
However upon using fix reaxff/species to calculate the number of individual species in the system, the total number of molecules in the system returns as ~1700. However the force field itself seems to accurately predict the number of molecules (upon running a very crude cluster analysis using the trajectory and reaxff/bonds output file, to count the total molecules in the system using OVITO)
Trying to use the “cutoff” modifier to apply a lower cut-off to all atoms does not seem possible since it does not accept “*” or group names are arguments.
While on this topic if there is anyone with expertise on this matter, the reaxff/species command outputs similar molecules with different numbers (eg. OLi - 35, OLi3 - 3, OLi6 -2), can I consider all of them similar. Why does the number arise in such cases?
I would appreciate any assistance in this regard, thanks!
I don’t understand this statement. It is most certainly possible to change the bond order cutoff. The cutoff parameter, however, requires providing the specific atom types that this applies to. There is nothing in the documentation that this keyword would accept wildcards. If something is not mentioned in the documentation, that means it is not implemented or supported.
In order to accept wildcard parameters the implementation of the command would have to be changed. Since ReaxFF based simulations usually do not have a lot of atom types, this is usually not much of a burden. Especially, since there is a default bond order cutoff with a reasonable value for most cases.
This means that fix reaxff/species has determined that there are 35 species containing one oxygen atom and one lithium atom, 3 species with one oxygen atom and 3 lithium atoms, and two species with one oxygen and six lithium atoms.
Fix reaxff/species applies the described algorithm and this is the outcome.
My question was whether it is possible to change the bond order cutoff for more than one atom type pair in a given command. Given this:
I see that it is not possible.
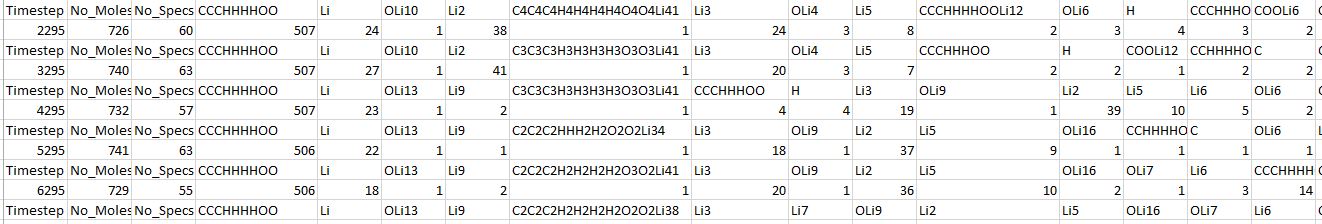
The issue with this is, if I consider the OLi6 as containing, one oxygen and 6 lithium atoms, the total number of Lithium ions in my system rises to bogus values. To shed more light, I’m providing further information regarding one such simulation, below.
The goal was to simulate the reduction of ethylene carbonate, and analyse the products formed from this reduction. Hence I prepared a system of 577 Ethylene carbonate molecules and 38 Li atoms. The data file was written using topotools, and I verified that the data file did indeed contain 38 Li atoms.
The fix reaxff/species command I used as part of the input script is as below:
fix ec_red all reaxff/species 10 10 1000 ec_reduction_prods.dat element C C C H H H H O O O Li
Here is a snippet from the “ec_reduction_prods.dat” file that was returned as an output from this simulation.
I assumed OLi10, OLi4 in this case are in fact similar, and can just be considered OLi. Otherwise, the number of atoms in my system don’t tally.
Please let me know if this simulation/result was entirely bogus, or if it was valid, then how do I interpret these results?
I would suspect that your counting is incorrect. Since the fix has determined 60 different species, it is quite likely that when manually tallying those numbers, some typo can happen.
The readability of your simulation output is negatively impacted by having multiple atom types for the same elements.
Also, your input file has a syntax error in the minimize command (1.0-15 instead of 1.0e-15) which was undetected until about December 2022 and thus requires using a later feature release version of LAMMPS or update3 of the stable release. In binaries without the bugfix, the number will be incorrectly interpreted as 1.0.
When starting the simulation, I see an extremely high pressure, which is worrisome and suggests that your initial geometry may rather far away from equilibrium.
Another problematic choice is the averaging over 100 steps (nevery*nrepeat) in fix reaxff/species. In combination with the default neighbor list settings this will lead to neighbor list rebuilds being delayed for 100 steps. At the (initial?) small timestep of 0.1 fs that may be acceptable, but may be problematic when using a larger timestep.
I also don’t quite understand your purpose of running fix npt first and only then raising the temperature to 800K.
You certainly need to discuss your simulation settings and equilibration protocol with your adviser/tutor/mentor or whoever else you are learning how to properly perform MD simulations from.
Finally, there also seems to be some rounding issue with assigning charges since there is a net charge of 0.577 which means exactly 0.001 per atom is added. This should be rectified, too.
Thank you for the suggestions, I’ll fix them in the future simulations.
What would you suggest to be the Nfreq I should use, to enable ideal re-neighbouring of atoms in the simulations.
That is very system specific. I would first run for a bit without fix reaxff/species (e.g. during equilibration) and use neigh_modify delay 0 every 1 check yes. Then after the run you can see how many neighbor list builds were done during the run. Divide the number of steps by this number, you have the average number of steps per neighbor list build (and make sure there are no dangerous builds, otherwise you need to do this step later in the equilibration). Since you have to switch to a fixed interval (i.e. check no), I would use something like 2/3rd of that number to be safe. You need to keep in mind that threshold for dangerous builds is deliberately low, so that if you have a small number of dangerous builds, it does not automatically mean you were missing neighbors, only that there is a growing probability the more dangerous builds there are.
Since in ReaxFF you need to use a rather short timestep, it is not unusual to have neighbor list rebuilds somewhat infrequent compared to when using bio force fields with fix shake to keep all bonds involving hydrogen atoms rigid. In the latter case, you may need to update neighbor list every few steps; with an aggressive timestep possibly even every step. That is not going to be the case here, once your system is properly equilibrated.