Dear all,
I used NVT and NPT ensemble to relax my structure. The graphical output of my dump file
looks good for NVT relaxation. However, my structures is totally shifted to a direction for NPT relaxation.
It is strange for me because a part of my structures goes outside of the pbc box and this finally leads to leakage of confined molecules at next steps.
Any comments will be appreciated.
This looks like you are using VMD. Is that correct?
Yes. I used VMD to visualize the output dump file. As I said everything looks Ok before using NPT ensemble. I used the p p p boundary condition.
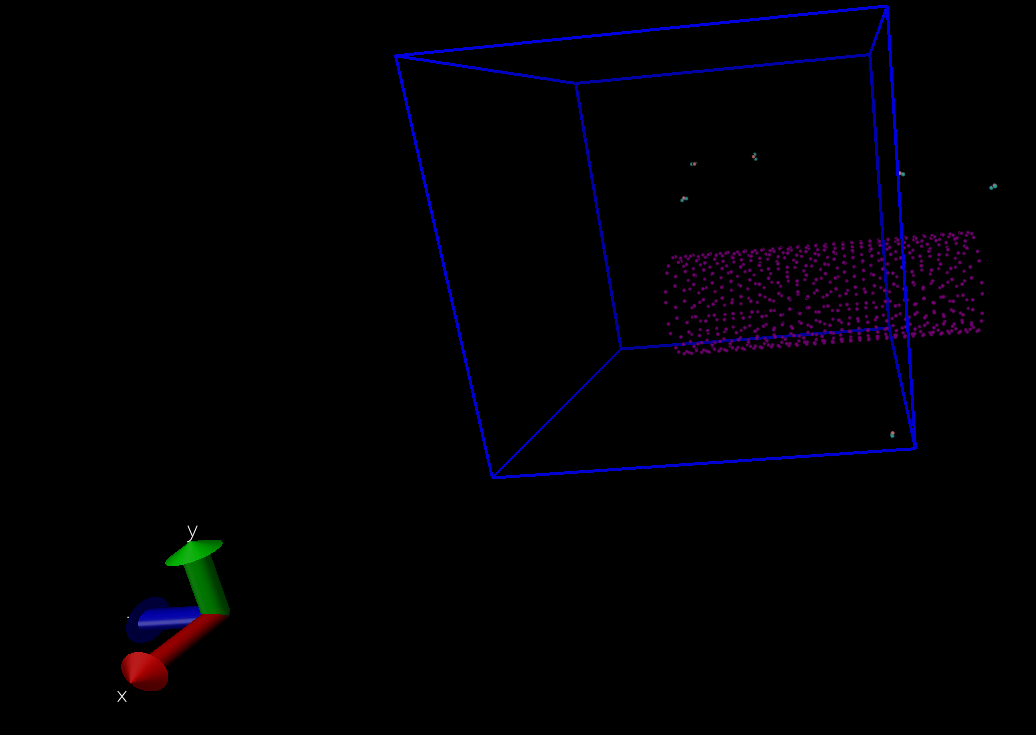
First, I thought that it was a problem with VMD. But I saw the leakage of my water molecules confined in the CNT at the following steps! This means the structure is really shifted during my NPT relaxation.
First off, VMD only reads the box dimensions, not the box origin, so when you draw a box it can show up anywhere and may not be commensurate with your system.
Second, whether your system is drifting or not, can only be seen, if you write out unwrapped coordinates or include the image flags in your dump file. Can you provide your input?
Or the top of the dump file (assuming it is a LAMMPS format text mode dump).
Since you have periodic boundaries (and the image looks like you did not write out image flags or unwrapped coordinates), the origin of your box is rather inconsequential. Whether your box is actually drifting can be best seen, if you monitor the center of mass velocity. E.g. with:
variable vcmx equal vcm(all,x)
variable vcmy equal vcm(all,y)
variable vcmz equal vcm(all,z)
and then adding v_vcmx, v_vcmy, and v_vcmz to the thermodynamic output.
It should fluctuate around zero.
You have to make certain, that there is no residual center of mass drift if you initialize velocities with the velocity command (see docs).
Using fix npt can have some “funny” effects on the system geometry, because will convert coordinates to fractional before changing the box and then converting back to regular.
Yes, it actually fluctuates around the zero! But, the interesting thing is about the box dimention which is:
0.2 39.7 xlo xhi
0.2 39.7 ylo yhi
0.2 41.7 zlo zhi
As you see, CM is not actualy locates at zero. This means, something extends the system and relocates the CM at zero point.
Yes, I attached the input file.
H2Omol.in (2.2 KB)
I used velocity create command, but I do not exactly know whether it is correct to call it before fix langavin or not!
In below figure, You can also see the lekage of the molecules after the system was completely relaxed with NVE ensemble and H2O molecules outside CNT were deleted.
Al in all, is there any commands to fix the structures coordinates? Is there any procedure to relocate the system after performing the npt ensemble?
I don’t understand what you mean by that. What is “CM”? Is it the center of mass (COM?).
The center of mass will be located wherever your initial geometry put it. It will rarely be at the origin.
The way the box change works is that LAMMPS will convert all positions to fractional coordinates (i.e. between 0 and 1), then update the box dimensions and the convert geometries back. The fixed point for the cell is then the center of the box and not the origin of the coordinate system. If you want to change that, you have to use the fixedpoint option of fix npt (see docs). Please also see the discussion for that keyword.
Why not? The crucial point is to use the “mom yes” option to the velocity command (see documentation).
I cannot comment on that without being able to look over your shoulder and do what an adviser usually does. I am quite certain that LAMMPS is operating correctly. As I already mentioned discussing the absolute box position in VMD is pointless since VMD doesn’t know it. With periodic boundaries and coordinate wrapping (since you only output x,y,z and not image flags or unwrapped coordinates) it may not be obvious what is “inside” and “outside”
I am convinced that this is mostly a question of visualization and a misunderstanding of what is happening.