Using lammps-3Mar20
I think my issue is with the way I am using the forcefield, but I hope this is the right place to ask this.
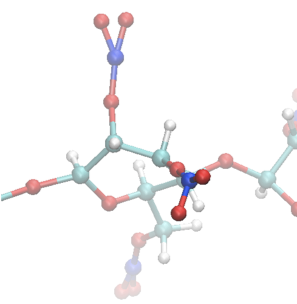
I am trying to run simulations on nitrocellulose. The calculations are running fine, mostly because I am mostly still using the examples from the moltemplate website.
However the ONO2 functional groups are not behaving as one would expect – the bond angles are closing up.
I think this is due to the descriptions I am choosing for the atoms, although I’ve been trying to correct this. I have been told that I can specify the angle_coeff, but I am not sure how to do this.
If anyone could help I would be very grateful
Thank you
Catriona Gibbon
dimer_nc.lt (8.05 KB)
oplsaa.lt (561 KB)
run.in.min (888 Bytes)
system.lt (521 Bytes)
Using lammps-3Mar20
I think my issue is with the way I am using the forcefield, but I hope this is the right place to ask this.
I am trying to run simulations on nitrocellulose. The calculations are running fine, mostly because I am mostly still using the examples from the moltemplate website.
However the ONO2 functional groups are not behaving as one would expect – the bond angles are closing up.
I think this is due to the descriptions I am choosing for the atoms, although I’ve been trying to correct this. I have been told that I can specify the angle_coeff, but I am not sure how to do this.
If anyone could help I would be very grateful
Thank you
Catriona Gibbon
(Sorry if this is a resubmission, but I haven’t heard anything back yet)
dimer_nc.lt (8.05 KB)
oplsaa.lt (561 KB)
run.in.min (888 Bytes)
system.lt (521 Bytes)
When you are not getting a reaction after a few days that usually means one of three things:
- people think this is a problem specific to a particular feature or LAMMPS-related software and want to let the author of that code respond
- people don’t know what to answer/recommend or are confused by what you are asking
- people think that this is something that you should know or figure out by yourself or discuss with your adviser/supervisor/colleagues that have more insight into the specifics of your research
In those cases it is rarely productive to repost the exact same text and files.
Axel.